
- Gösterim: 22012
Keratoderma ve Palmoplantar Keratoderma (PPK)
Keratoderma, derinin nasırlaşması ve kalınlaşması olarak tanımlanan klinik bir durumdur. Özellikle avuç içi ve ayak tabanlarında görülen bu duruma palmoplantar keratoderma (PPK) adı verilir. Genetik geçişli formları ise "kalıtsal palmoplantar keratodermalar" veya "hereditary palmoplantar keratoderma" olarak bilinir. Kalıtsal PPK'lar, klinik özellikleri ve tutulum dereceleri bakımından çeşitlilik gösteren geniş bir hastalık grubudur. Bu durum, el ve ayaklarda görünüş veya fonksiyon bozukluklarına (mutilasyon) yol açabilir. Ayrıca saç, tırnak, diş ve terleme fonksiyonlarında bozukluklar gibi sistemik belirtilerle de birlikte görülebilir. Hastalığın çoğu formu bebeklik döneminde fark edilebilir ve tırnak, diş veya diğer organ anomalileriyle birlikte ortaya çıkabilir. Bu nedenle, kalıtsal PPK'ların erken tanısı, klinik bulgular ve moleküler çalışmalar ışığında alt tiplerin belirlenmesi açısından son derece önemlidir.
PPK'nın ortaya çıkmasındaki moleküler mekanizma son yıllarda daha iyi anlaşılmıştır. Cildin keratinizasyon sürecinde rol oynayan proteinleri kodlayan genlerdeki mutasyonlar bu durumdan sorumludur. Ne yazık ki, günümüzde bu hastalık grubu için radikal ve kalıcı bir tedavi henüz bulunmamaktadır.
Avuç içi ve ayak tabanlarındaki cilt, diğer bölgelerden farklı ve benzersiz özelliklere sahiptir. Bu bölgelerde parmak izleri (dermatoglifikler) bulunur, yoğun ter bezleri olmasına karşın yağ bezleri ve kıllar yoktur. Ayrıca, bu alanlardaki keratinizasyon (cilt hücrelerinin çoğalması ve farklılaşması) diğer bölgelerden farklıdır. Bu özellikler, palmoplantar keratodermanın (PPK) neden özellikle el ve ayaklarda görüldüğünü ve hastalığın karakteristik belirtilerini açıklar.
Tanısal Yaklaşım
PPK tanısı için hastadan ayrıntılı bir tıbbi ve mesleki öykü alınması önemlidir. Bu, edinilmiş PPK'nın olası nedenlerini ayırt etmeye yardımcı olur. Bu nedenler şunları içerebilir:
-
Mesleki Faktörler: Yoğun el emeği gerektiren meslekler.
-
Cilt Hastalıkları: Kronik kontakt dermatit, atopik dermatit, sedef hastalığı.
-
Enfeksiyonlar: HIV enfeksiyonu.
-
Paraneoplastik ve Metabolik Bozukluklar: Hipotiroidizm, miksödem.
-
Kimyasallara Maruz Kalma: Arsenik, hidrokarbonlar.
-
İlaçlar: Lityum, kemoterapötik ilaçlar.
Bu bilgilerin toplanması, hastalığın temelinde yatan nedeni belirlemek ve doğru tanıya ulaşmak için önemlidir.
Cildin keratinizasyon sürecinde rol oynayan genlerdeki mutasyonlar, iktiyoz ve palmoplantar keratoderma (PPK) gibi hastalıklara yol açar. Bu mutasyonlar, keratinizasyonun bozulmasına ve cildin en dış katmanı olan stratum korneumun aşırı kalınlaşmasına neden olur.
Keratinler, hücrelerin yapısal bütünlüğünü sağlayan proteinlerdir. Avuç içi ve ayak tabanındaki cildin katmanlarında farklı keratin tipleri ifade edilir:
-
Bazal katmanda: K5 ve K14
-
Suprabazal katmanda: K1, K9, K6a/K16 ve K6b/K17
Keratin genlerindeki mutasyonlar, mutasyonun konumuna göre değişen şiddette PPK'ya yol açar. Keratin peptitlerinin korunmuş bölgelerindeki mutasyonlar şiddetli PPK ile sonuçlanırken, daha değişken bölgelerdeki mutasyonlar daha hafif klinik belirtilere neden olur.
Hücreler arası yapışmayı ve iletişimi sağlayan proteinlerdeki mutasyonlar da PPK gelişiminde rol oynar:
-
Desmozomlar: Keratin filamentlerini hücre zarına bağlayan ve keratinositler arasında güçlü bir yapışma sağlayan bağlantı noktalarıdır. PPK'lı hastalarda desmozomlar daha büyüktür ve bu proteinlerdeki mutasyonlar PPK'ya neden olabilir.
-
Lorikrin: Stratum korneumda kornifiye hücre zarfının oluşumunda kritik bir rol oynar. Bu proteindeki mutasyonlar, anormal kornifikasyona ve hücre ölümüne yol açarak hafif iktiyoziform kızarıklığa ve PPK'ya neden olur.
-
Gap Junction (Boşluk Bağlantısı) Proteinleri: Konneksin proteinlerinden oluşan bu kanallar, hücreler arası iletişimi sağlar. Konneksin mutasyonları, anormal protein birikimine ve dolayısıyla keratodermaya yol açar.
Bu proteinlerin yanı sıra, sistein lizozomal proteaz, geçici reseptör potansiyeli vaniloid 3, salgılanan LY6/ürokinaz tipi plazminojen aktivatör reseptörü (uPAR) ile ilişkili protein 1 gibi çeşitli diğer moleküller de PPK gelişiminde rol oynar.
Palmoplantar Keratoderma (PPK) Klinik Tipleri
Palmoplantar keratoderma, klinik olarak farklı şekillerde kendini gösterebilir:
-
Diffüz Tip: En sık görülen PPK çeşididir. Avuç içi ve ayak tabanlarının tamamını kapsayan, pürüzsüz ve mumsu bir kalınlaşma ile karakterizedir.
-
Fokal Tip: Sadece belirli alanları etkiler.
-
Çizgisel veya Noktasal Tip: Ciltte çizgi veya noktalar şeklinde kendini gösterir.
Diffüz PPK Alt Tipleri: Diffüz PPK, avuç içi ve ayak tabanlarından komşu bölgelere yayıldığında "Transgrediensli diffüz palmoplantar keratoderma" olarak tanımlanır. Bu yayılım; ellere, ayaklara, bileklerin iç kısımlarına ve aşil tendonlarına kadar uzanabilir. Ayrıca, bazı PPK formları yaşla birlikte ilerler veya iyileşir. Bu durum "Progrediens palmoplantar keratoderma" olarak adlandırılır.
PPK'nın sınıflandırılması, hastanın klinik özelliklerine, genetik geçişine, moleküler mutasyona, diğer dermatolojik bulgulara ve sistemik tutuluma göre yapılır.
Mutilasyonun Olmadığı veya Az Görüldüğü Diffüz PPK Tipleri
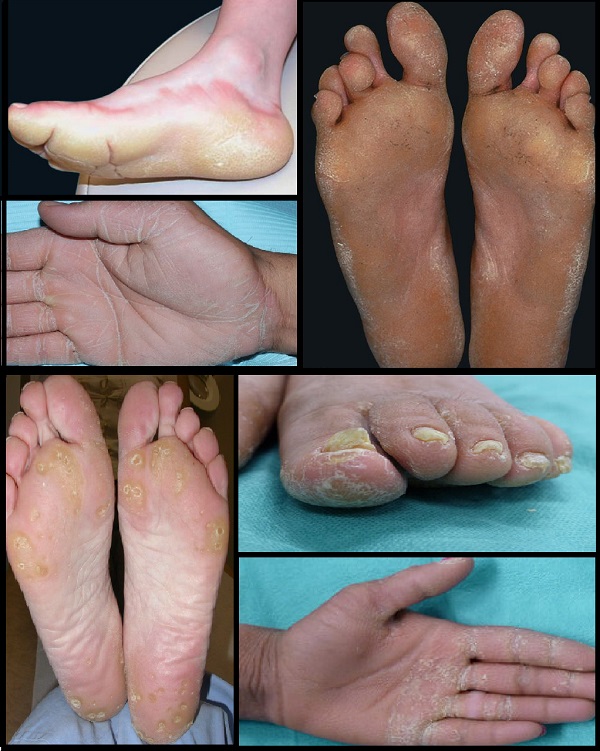
- Diffüz tutulum gösteren, mutilasyonun olmadığı veya az görüldüğü palmoplantar keratoderma (PPK), avuç içlerinin ve ayak tabanlarının tamamında kalınlaşma ile karakterize bir durumdur. Bu tipte, parmaklarda veya el-ayak yapısında ciddi bozulmalar görülmez veya nadirdir.
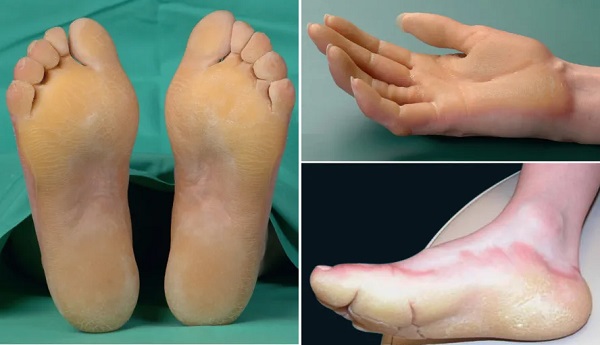
Bu guruptaki keratodermalar;
-
- Vörner (Unna-Thost) Palmoplantar Keratoderma: Klinik: Avuç içi ve ayak tabanında yaygın (diffüz) kalınlaşma. Genetik: KRT1 ve KRT2 gen mutasyonları, otozomal dominant geçiş.Özellikler: Çevresel yayılım (transgrediens yok), tırnak ve terleme bozukluğu (hiperhidrozis) yok. Bebeklikte nadiren su toplaması görülür, ciddi bozulma (mutilasyon) meydana gelmez.
- DSG1 Mutasyonları ile Palmoplantar Keratoderma: Klinik: Avuç içi ve ayak tabanında diffüz kalınlaşma.Genetik: DSG1 gen mutasyonu, otozomal dominant geçiş.Özellikler: Transgrediens yayılım yok. Tırnaklarda hafif sararma ve ayrılma (onikoliz) olabilir. Hiperhidrozis ve mutilasyon yok.
- Nagaşima Palmoplantar Keratoderma: Klinik: Avuç içi ve ayak tabanında hafif kalınlaşma ve kızarıklık. Genetik: SERPINB7 gen mutasyonu, otozomal resesif geçiş. Özellikler: Diz, dirsek ve aşil tendonu bölgelerine yayılan transgrediens yayılım var. Suya maruz kalınca beyaz süngerimsi bir görünüm ve kötü koku oluşur. Tırnak tutulumu yoktur ancak hiperhidrozis vardır.
- Botni Palmoplantar Keratoderma: Klinik: Avuç içi ve ayak tabanında kalın diffüz kalınlaşma. Genetik: AQP5 gen mutasyonu, otozomal dominant geçiş. Özellikler: Transgrediens yayılım ve hiperhidrozis vardır. Tırnaklar pürüzlüdür. Suya maruz kalınca beyaz süngerimsi bir görünüm oluşur.
- Greither'in Palmoplantar Keratoderması: Klinik: Avuç içi ve ayak tabanında diffüz kalınlaşma. Genetik: KRT1 gen mutasyonu, otozomal dominant geçiş. Özellikler: Transgrediens yayılım ve hiperhidrozis vardır. Bebeklikte başlar, geçici su toplamaları görülebilir ve ilerleyicidir. 40-50 yaşlarında gerileyebilir. Mutilasyon meydana gelir.
- Sybert Sendromu: Klinik: Avuç içi ve ayak tabanında diffüz kalınlaşma, göz çevresinde ve ağız kenarlarında kızarıklık (periorbital ve perioral eritem). Genetik: Gen mutasyonu bilinmiyor, otozomal dominant geçiş. Özellikler: Diz ve dirseklere yayılan transgrediens yayılım var. Tırnak tutulumu ve hiperhidrozis yok. Mutilasyon meydana gelir.
- Gamborg Nielsen Palmoplantar Keratoderması: Klinik: Avuç içi ve ayak tabanında diffüz kalınlaşma. Genetik: SLURP1 gen mutasyonu, otozomal resesif geçiş.Özellikler: El sırtlarına yayılan transgrediens yayılım ve parmak boğumlarında kalınlaşma (knuckle pad) gelişir. Tırnak tutulumu, hiperhidrozis ve mutilasyon yok.
- Akral Keratoderma: Klinik: Avuç içi ve ayak tabanında diffüz veya çizgisel kalınlaşma. Genetik: Gen mutasyonu bilinmiyor, otozomal resesif geçiş. Özellikler: Diz, dirsek ve aşil tendonuna yayılan transgrediens yayılım var. Tırnak tutulumu ve hiperhidrozis yok. Mutilasyon görülebilir.
- Huriez Sendromu: Klinik: Avuç içi ve ayak tabanında diffüz kalınlaşma. Genetik: Gen mutasyonu bilinmiyor, otozomal dominant geçiş. Özellikler: Ellerde ve ayaklarda parşömen benzeri kuru kalınlaşma ve büyüme geriliği görülür. Tırnaklarda şekil bozukluğu veya gelişim bozukluğu (distrofi/hipoplazi) vardır. Yassı hücreli karsinom (SCC) riski artmıştır. Hiperhidrozis yoktur.
- KID (Keratitis, İktiyozis, Sağırlık) Sendromu: Klinik: Avuç içi ve ayak tabanında diffüz kalınlaşma. Genetik: GJB2 (GJB6) gen mutasyonu, otozomal dominant geçiş. Özellikler: Yenidoğanlarda iktiyoziform kızarıklık, saç dökülmesi (alopesi) ve siğil benzeri kalınlaşmalar (akrofasiyal verrukoz hiperkeratoz) görülür. Ayrıca sensörinöral işitme kaybı, gözde keratit ve tekrarlayan bakteriyel enfeksiyonlar mevcuttur. SCC riski artmıştır. Tırnaklarda distrofi vardır ve hiperhidrozis yoktur.
- Bart–Pumphrey Sendromu: Klinik: Avuç içi ve ayak tabanında diffüz, fokal veya noktasal kalınlaşma. Genetik: GJB2 gen mutasyonu, otozomal dominant geçiş. Özellikler: Tırnaklarda beyaz lekeler (lökonişya) görülür. Sensörinöral işitme kaybı, parmak boğumlarında kalınlaşma (knuckle pad) ve meme ile koltuk altında kistler oluşur. Hiperhidrozis yoktur.
- Howel–Evans Sendromu: Klinik: Avuç içi ve ayak tabanında sarımsı, nasır benzeri diffüz kalınlaşma. Genetik: RHBDF2 gen mutasyonu, otozomal dominant geçiş. Özellikler: Transgrediens yayılım yok. Tırnak tutulumu ve hiperhidrozis yoktur. Keratozis pilaris, el ve ayak derisinde çatlaklar ve enfeksiyonlar görülür. Kronik rinit, maksiller kemikte kireçlenme kaybı, diş alveollerinde erime ve diş kaybı, ağız içinde lökoplaki ve ilerleyen yaşlarda yemek borusu karsinoması riski mevcuttur.
- Diffüz tutulum gösteren ve mutilasyonlar gösteren palmoplantar keratodermalar: Mutilasyonun ve şiddetli klinik belirtilerin görüldüğü palmoplantar keratodermalar;
- Mal de Meleda (Gamborg – Nielsen / Norrbotten) Palmoplantar Keratoderma: Klinik: Avuç içi ve ayak tabanında şiddetli, yaygın (diffüz) kalınlaşma. Genetik: SLURP1 gen mutasyonu, otozomal resesif geçiş. Özellikler: Tırnaklarda şiddetli bozukluklar (distrofiler) ve aşırı terleme (hiperhidrozis) görülür. Dirsekler, dizler ve aşil tendonu bölgelerine yayılan transgrediens yayılım vardır. El ve ayak derisinde çatlaklar, kötü koku ve enfeksiyonlar sıktır. Parmaklarda incelme, eklem sertliği ve kontraktürler, hatta spontan parmak ampütasyonları gibi ciddi mutilasyonlar meydana gelir.
- Vohwinkel Sendromu: Klinik: Avuç içi ve ayak tabanında şiddetli, sarımsı diffüz kalınlaşma. Genetik: GJB2 gen mutasyonu, otozomal dominant geçiş. Özellikler: Tırnak distrofileri ve hiperhidrozis mevcuttur. Eklemlerde ve bileklerde denizyıldızı şekilli kalınlaşmalar (keratozlar) ile transgrediens yayılım görülür. Ayrıca sensörinöral sağırlık, saç dökülmesi (alopesi), kas zayıflığı (miyopati) ve odaklanmış epilepsi gibi sistemik belirtiler görülebilir.
- İhtiyosiform Vohwinkel Sendromu (Camisa Hastalığı): Klinik: Avuç içi ve ayak tabanında şiddetli, sarımsı diffüz kalınlaşma. Genetik: LOR (Lorikrin) gen mutasyonu, otozomal dominant geçiş. Özellikler: Tırnak distrofileri ve hiperhidrozis vardır. Transgrediens yayılım ve hafif iktiyozis görülür. Parmaklarda incelme (psödoainhum) ve odaklanmış epilepsi görülebilir. Sensörinöral sağırlık yoktur.
- Olmsted Sendromu: Klinik: Avuç içi ve ayak tabanında şiddetli, sarı-kahverengi diffüz kalınlaşma ve çatlaklar. Genetik: TRPV3 gen mutasyonu, otozomal dominant geçiş. Özellikler: Tırnak distrofileri ve aşırı veya hiç terleme (hiperhidroz/anhidroz) görülebilir. Ağız, burun, göz gibi açıklıkların çevresinde, boyunda, göğüste, karın ve kasık kıvrımlarında, uyluk, dirsek ve dizlerde kalınlaşma (periorifisyal hiperkeratoz) mevcuttur. Saç dökülmesi, enfeksiyonlar, ağızda beyaz lekeler (oral lökokeratoz), kronik göz kapağı iltihabı (blefarit), kornea bozuklukları, büyüme geriliği, yüksek frekanslı işitme kaybı, anormal diş yapısı ve kemik deformiteleri gibi çok sayıda sistemik belirti görülür. Kötü koku da eşlik edebilir.
- KLICK Sendromu (İktiyozisli Keratozis Linearis): Klinik: Avuç içi ve ayak tabanında şiddetli diffüz kalınlaşma. Genetik: POMP gen mutasyonu, otozomal resesif geçiş. Özellikler: Kol fleksörleri ve bilekler üzerinde çizgisel düzende kalınlaşmış plaklar (hiperkeratoz plakları), iktiyozis ve papüller ile birlikte transgrediens yayılım görülür.
- Periodontitis ile birlikte Palmoplantar Keratoderma (Papillon-Lefèvre Sendromu): Klinik: Sıklıkla şiddetli diffüz, nadiren noktasal veya çizgisel kalınlaşma. Ayak tabanları avuç içlerinden daha şiddetli etkilenir. Kötü kokulu hiperhidroz sıktır. Genetik: CTSC (katepsin C) gen mutasyonu, otozomal resesif geçiş. Özellikler: Doğumda veya erken bebeklikte avuç içi ve ayak tabanında kızarıklıkla başlar. Parmak eklemleri, dirsekler, dizler, aşil tendonu ve bileklerde psoriasiform plaklar görülür. Hem süt hem de kalıcı dişleri etkileyen şiddetli periodontitis ve diş eti iltihabı nedeniyle dişler erken kaybedilir, 14-15 yaşına gelindiğinde dişsiz kalınır. Genellikle Staphylococcus aureus'a bağlı cilt ve karaciğer enfeksiyonları, hafif zihinsel gerilik, beyin dokusunda ve koroid pleksusta anormal kalsiyum birikimi de bildirilmiştir.
- Fokal tutulum gösteren palmoplantar keratodermalar

-
- Palmoplantar Keratoderma Nummularis: Klinik: Avuç içi ve ayak tabanında madeni para şeklinde, nasıra benzeyen kalınlaşmalar (keratodermalar). Genetik: KRT16, KRT6c, DSG1 veya TRPV3 gen mutasyonları, otozomal dominant geçiş. Özellikler: Transgrediens yayılım yok. Ayak tırnaklarında kalınlaşma (hipertrofi) görülebilir ancak genellikle tırnak tutulumu yoktur. Hiperhidrozis mevcuttur. Çocukluk çağında başlar, mekanik stresle tetiklenebilir ve ağrılı hale gelebilir. Kıl köklerinde kalınlaşma (foliküler hiperkeratoz) ve ayak tabanında kabarcıklar (vezikülasyon) görülebilir. Mutilasyon (yapısal bozulma) yoktur. Ağız veya cinsel organ çevresinde minimal beyaz lekelenmeler (lökokeratoz) görülebilir.
- Pachyonychia Congenita (Konjenital Pakinonişi): Klinik: Ayak tabanında kalınlaşma (plantar keratoderma), taban ağrısı ve kalınlaşmış ayak tırnakları en temel özelliklerdir. Hastaların %90'ından fazlasında 5 yaşından önce gelişir. Tırnak bozukluğu ortalama 2.8 yaşında başlar. Keratodermaya eşlik eden çatlaklar ve ülserasyonlar olabilir. Ayak tabanı ağrısı yaşam kalitesini ciddi şekilde etkiler. Genetik: KRT6A, KRT6B, KRT16 veya KRT17 genlerindeki mutasyonlar, otozomal dominant geçiş. Özellikler: Cilt, tırnaklar, ağız mukozası ve dişleri etkileyen sistemik bir hastalıktır.
- Yün Saçlar + Aritmi Yapan Kardiyomiyopati + Striata PPK: Klinik: "Yün saçlar" adı verilen kıvırcık saçlar, aritmi ile seyreden bir kardiyomiyopati ve avuç içi/ayak tabanında çizgisel (striata) formda keratoderma görülür. Bu, bir sendromun üç ana bileşenidir.
- Çizgi şeklinde, linear tutulum gösteren palmoplantar keratodermalar
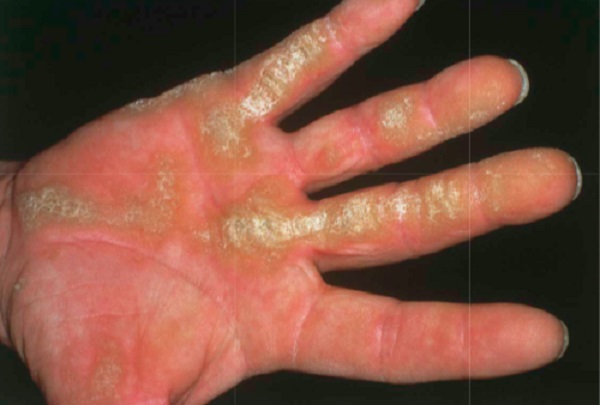
-
Çizgisel (Linear) palmoplantar keratodermalar, avuç içleri ve ayak tabanlarında, özellikle parmakların fleksör yüzeyleri ve baskı noktalarında çizgisel şekilde kalınlaşmalarla karakterizedir. Brünauer–Fuhs–Siemens sendromu olarak bilinen Tip I, Tip II ve Tip III olmak üzere 3 klinik alt tipi bulunur. DSG1 (Desmoglein 1), DSP (Desmoplakin) ve KRT1 (Keratin 1) genlerindeki mutasyonlardan kaynaklanır ve otozomal dominant geçişlidir. Bu tiplerde çevresel yayılma (transgrediens) yoktur. Tırnak tutulumu görülmez. Tip I'de hiperhidrozis (aşırı terleme) mevcuttur. Nadiren diz ve dirseklerde kalınlaşma (hiperkeratoz) ve kötü koku görülebilir. Tip I'de ayak tabanında ağrıya neden olabilir. Hastalık yaşla birlikte iyileşme gösterebilir ve kendiliğinden uzuv kaybı (spontan ampütasyon), yani mutilasyon görülmez.
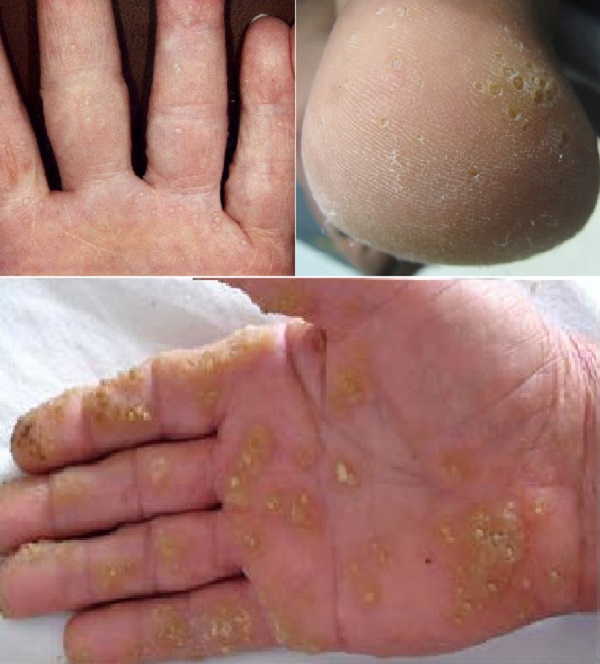
- Noktalar şeklinde, punktate tutulum gösteren palmoplantar keratodermalar; avuç içi ve ayak tabanında nokta şeklinde kalınlaşmalarla karakterizedir.
- Tip-IA (Buschke–Fischer–Brauer Sendromu): Klinik: Avuç içi ve ayak tabanında nokta şeklinde kalınlaşmalar (keratodermalar). Genetik: AAGAB gen mutasyonu, otozomal dominant geçiş. Özellikler: Çevresel yayılma (transgrediens) ve tırnak tutulumu yoktur. Hiperhidrozis (aşırı terleme) görülmez. Genellikle geç çocukluk veya ergenlik döneminde başlar. Yaş ilerledikçe kalınlaşmaların sayısı ve boyutu artar. Suya maruz kaldığında kötüleşir. Yapısal bozulma (mutilasyon) yoktur.
- Tip-IB: Klinik: Klinik özellikleri Tip-IA ile aynıdır. Genetik: Tip-IB'nin tek farkı, gen mutasyonunun farklı olmasıdır. Diğer tüm klinik özellikler aynıdır.
- Tip II (Porokeratotik Tip): Klinik: Avuç içi ve ayak tabanında nokta şeklinde kalınlaşmalar ve keratoz tıkaçlı çukurlar görülür. Genetik: Gen mutasyonu bilinmiyor, otozomal dominant geçiş. Özellikler: Transgrediens yayılım ve tırnak tutulumu yoktur. Hiperhidrozis görülmez. Yirmili yaşların sonunda başlar ve erkeklerde yağ bezi büyümesi (sebase hiperplazi) görülebilir. Mutilasyon yoktur.
- Tip-III (Akrokeratoelastoidozis): Klinik: Avuç içi ve ayak kenarlarında keratotik papüller (sert, kabartılı döküntüler) mevcuttur. Genetik: Gen mutasyonu bilinmiyor, otozomal dominant geçiş. Özellikler: Nadiren çevresel yayılma veya tırnak tutulumu görülür. Hiperhidrozis yoktur. Ergenlik veya erişkinlik döneminde başlar. Mutilasyon görülmez.
Palmoplantar Keratodermanın (PPK) İlişkili Olduğu Durumlar
Palmoplantar keratoderma (PPK), bazen tek başına bir durum olarak değil, farklı deri hastalıkları ve sendromlarla ilişkili olarak ortaya çıkabilir. Bu nedenle, klinik değerlendirme sırasında şunlar gibi durumların varlığına dikkat edilmelidir:
-
Eritrokeratoderma variabilis
-
Pityriazis rubra pilaris
-
Doğuştan iktiyozis
-
Epidermolizis bülloza
-
Darier hastalığı
-
Akrokeratozis verruciformis
-
Cowden hastalığı
-
Bazal hücreli nevüs sendromu (Gorlin sendromu)
-
Akrokeratoelastoidozis
-
Tirozinemi tip II
-
Kistik fibrozis
PPK ayrıca kalp kusurları, işitme kusurları, nöropatiler, göz kusurları ve özofagus veya cilt kanseri riskinin artması gibi sistemik hastalıkların bir belirtisi olarak da ortaya çıkabilir.
Tanısal Yaklaşım
Kalıtsal PPK'lar, geniş bir genetik ve klinik yelpazeye sahiptir. Bu nedenle, sadece klinik belirtilere dayanarak doğru bir tanıya ulaşmak zordur. Tanı sürecinde, keratinizasyondan sorumlu proteinlerin incelenmesi veya deri biyopsisi gibi patolojik tanımlamalar kritik öneme sahiptir.
Ancak, klinik modellerin oluşturulması ve her modelin detaylı tanımlanması, doğru tanıyı kolaylaştırabilir. Bu modeller, hastanın aşağıdaki özelliklerine odaklanır:
-
Keratodermanın klinik görünümü ve tutulum derecesi.
-
El, ayak ve parmaklarda meydana gelen mutilasyon (yapısal bozulma) varlığı.
-
Saç, tırnak, diş ve terleme fonksiyon bozuklukları gibi eşlik eden belirtiler.
-
Sistemik tutulumun olup olmadığı.
Yüksek maliyetine rağmen, moleküler çalışmalar bu farklı klinik formları doğru bir şekilde belirlemek için son derece önemlidir.
Genetik Danışmanlık
PPK'ların çoğu otozomal dominant veya resesif olarak genetik geçiş gösterir. Bu nedenle, hastalar ve aileleri için genetik danışmanlık hayati öneme sahiptir. Özellikle, resesif genetik varyantları değerlendirmek için ebeveynlerde akrabalık öyküsünün sorgulanması önemlidir.
Tedavi
Kalıtsal palmoplantar keratodermalar (PPK) yaşam boyu süren ve yaşam kalitesini ciddi şekilde etkileyen hastalıklardır. Ne yazık ki, günümüzde bu durum için spesifik ve kalıcı bir tedavi bulunmamaktadır. Tedavinin amacı semptomları hafifletmek ve hastanın yaşam kalitesini artırmaktır.
Cilt Bakımı ve Topikal Tedaviler
-
Hasta Eğitimi: Hastaların hiperkeratotik el ve ayak bakımı konusunda eğitilmesi önemlidir.
-
Mekanik Debridman: Kalınlaşmış bölgelerin mekanik olarak temizlenmesi geçici rahatlama sağlar.
-
Topikal Keratolitikler: Bol miktarda kullanılan topikal keratolitik ajanlar, cildin kalınlığını azaltarak yardımcı olabilir.
Sistemik Tedaviler
-
Sistemik Retinoidler: Çoğu PPK hastasında hiperkeratoz belirtilerini iyileştirebilir. Ancak bazı keratoderma tiplerinde dikkatli kullanılmalıdır. Örneğin, Papillon-Lefèvre sendromunda hem keratoderma hem de diş eti iltihabında belirgin bir iyileşme sağlasa da, epidermolitik palmoplantar keratodermada ciltte soyulmayı (epidermoliz) tetikleyebilir.
-
D Vitamini: Kalıtsal PPK tedavisinde etkili olduğu bildirilmiştir.
-
Erlotinib: Epidermal büyüme faktörü inhibitörü olan bu ilaç, önemli yan etkiler olmaksızın PPK'yı hafifletebilir.
-
Topikal Gentamisin: Nagashima tipi palmoplantar keratozun tedavisinde kullanılabilir.
Bu tedavilerin yanı sıra, ikincil enfeksiyonlara karşı dikkatli olunmalı ve gerektiğinde uygun tedaviler uygulanmalıdır.