
- Gösterim: 19446
Albinizm, göz, deri ve saçın pigmentasyonu olan melaninin biyosentezindeki bozukluklardan kaynaklanan, kalıtsal bir hastalık grubudur. Dünya genelinde yaklaşık 17.000 doğumda bir görülür.

Türleri
Albinizm, etkilenen dokulara göre iki ana gruba ayrılır:
-
Oküler Albinizm (OA): Sadece gözlerin etkilendiği türdür.
-
Okülokütanöz Albinizm (OKA): Göz, saç ve derinin de etkilendiği türdür.
Göz Sistemindeki Değişiklikler
Albinizmde göz bulguları tanı için son derece önemlidir ve hem OA hem de OKA'nın tüm tiplerinde görülür. Belirtiler şiddetine göre değişiklik gösterebilir:
-
Nistagmus: Gözlerin istemsiz, hızlı hareketleri.
-
Fotofobi: Işığa karşı aşırı duyarlılık.
-
Hipopigmentasyon: İris ve retinal epitelde pigment azlığı.
-
Foveal Hipoplazi: Net görmeden sorumlu fovea bölgesinin tam olarak gelişmemesi.
-
Kırma Kusurları: Miyopi, hipermetropi veya astigmatizm gibi görme bozuklukları.
-
Görme Keskinliğinde Azalma: Azalmış görme yeteneği.
-
Optik Sistem Anomalileri: En temel değişiklik, optik sinirlerin yanlış bir rotada ilerlemesi ve optik kiazmada aşırı çaprazlaşma olmasıdır. Bu durum, şaşılık ve azalmış derinlik algısı (stereoskopik görme) ile sonuçlanır.
OKA'nın tüm tiplerinde görülen azalmış görme keskinliği ve oküler nistagmus, bu durumu doğuştan gelen diğer hipopigmentasyon bozukluklarından ayırmada kritik özelliklerdir.
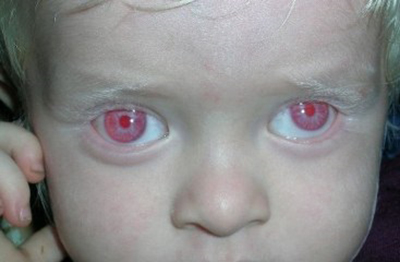
Albinizm, okülokütanöz albinizm (OKA) ve oküler albinizm (OA) olmak üzere iki ana tipe ayrılır ve her ikisinin de farklı alt tipleri vardır. Pigment yokluğu her tipte farklılık gösterir. En şiddetli tip olan OKA1A'da yaşam boyu melanin üretimi tamamen yokken, diğer tiplerde (OKA1B, OKA2, OKA3 ve OKA4) zamanla bir miktar pigment birikimi görülebilir.
Okülokütanöz Albinizm (OKA) Alt Tipleri
-
OKA 1: TYR (tirozin) genindeki mutasyonlar sonucu oluşur ve otozomal resesif olarak kalıtılır.
-
OKA 1A (En Şiddetli Form): Tirozinaz enzimi tamamen yoktur. Saç, kaş, kirpik ve deri bembeyazdır. İrisler açık renkli ve tamamen saydamdır. Görme keskinliği çok azdır ve ışığa karşı aşırı duyarlılık (fotofobi) şiddetlidir. Pigment gelişmez ve belirtiler yaşla değişmez.
-
OKA 1B (Sarı Mutant Albinizm): Tirozinaz aktivitesi azalmıştır. Yıllar içinde hastanın saçları ve cildi hafifçe koyulaşır.
-
OKA TS (Sıcaklığa Duyarlı): Tirozinaz enzimi 35°C altında fonksiyon gösterebilir. Bu nedenle, yaş ilerledikçe el ve ayaklar gibi soğuk bölgelerde bir miktar pigmentasyon gözlenebilir.
-
-
OKA 2: P genindeki mutasyonlar sorumludur. Pigmentasyon normalden hiç olmayana kadar değişebilir. Yenidoğanlarda genellikle pigmentli saç bulunur. Nevus (ben) ve çiller yaygındır. Göz rengi değişkenlik gösterir ve OKA1A'daki pembe gözler genellikle görülmez. Görme keskinliği genellikle OKA1'den daha iyidir.
-
OKA 3 (Rufous Albinizm): TRP-1 gen mutasyonu vardır. Sadece siyahi ırkta tanımlanmıştır ve kırmızı saçlı, kırmızımsı kahverengi ciltli bireylerde görülür.
-
OKA 4: Yeni tanımlanan bir tiptir. Sorumlu genin membranla ilişkili bir taşıyıcı protein olduğu düşünülse de işlevi tam olarak bilinmemektedir. Klinik bulgular açısından OKA2'den ayırt edilmesi zor olabilir.
Oküler Albinizm (OA)
-
Kalıtım ve Tanı: X'e bağlı geçiş gösterir. Taşıyıcı kadınların göz dibinde "çamur sıçramış fundus" görünümü vardır. Tanı, deri biyopsisinde makromelanozomların görülmesiyle konur.
Tanı, Tedavi ve Yönetim
-
Tanı: OKA'da tanı, karakteristik göz semptomları ve saç-deri pigmentasyonundaki azalmanın klinik bulgularıyla konur. Patolojik incelemede, melanosit sayısı normal olmasına rağmen, melanin içeriği azalmıştır veya yoktur.
-
Prenatal Tanı: OKA, gebeliğin 10-12. haftalarında koryonik villus örneklemesi veya amniyosentez ile elde edilen DNA kullanılarak prenatal olarak teşhis edilebilir.
-
Tedavi ve Korunma: Albinizmin primer tedavisi yoktur. Fotofobi (ışık duyarlılığı) ve deri kanseri riski nedeniyle hastaların güneşten koruyucu kremler ve güneş gözlüğü kullanması hayati önem taşır.
Albinizm ile İlişkili Sendromlar
Albinizm, bazen genetik bir bozukluğun veya sendromun parçası olarak ortaya çıkabilir. Bu durumlar, pigmentasyon sorunlarının yanı sıra, bağışıklık sistemi, nörolojik fonksiyonlar ve diğer organ sistemlerini de etkileyebilir.
Chediak-Higashi Sendromu (CHS)
Chediak-Higashi Sendromu (CHS), çocuklarda nadir görülen, otozomal resesif geçişli bir hastalıktır. LYST veya CHS1 genindeki mutasyondan kaynaklanır.
-
Belirtileri:
-
Okülokütanöz albinizm: Hastaların saç rengi açık, gümüşsü renkten koyu kahverengiye kadar değişebilir. Işığa karşı aşırı duyarlılık (fotofobi) görülür.
-
Bağışıklık Yetmezliği: Nötrofil sayısında azalma (nötropeni), bağışıklık hücrelerinin fonksiyon bozukluğu ve tekrarlayan enfeksiyonlar görülür.
-
Kanama Eğilimi: Trombosit eksikliğine bağlı kanamaya yatkınlık mevcuttur.
-
-
Seyri: Hastaların büyük çoğunluğu (%90) ilk 10 yılda enfeksiyon veya kanama gibi komplikasyonlar nedeniyle hayatını kaybeder. Hayatta kalanlarda ise ilerleyici nörolojik sorunlar gelişir.
Hermansky-Pudlak Sendromu (HPS)
Hermansky-Pudlak Sendromu (HPS), otozomal resesif geçişli genetik bir hastalıktır. Okülokütanöz albinizm, kanama yatkınlığı ve çeşitli organlarda seroid lipofüksin birikimi ile karakterizedir.
-
Belirtileri:
-
Albinizm: Deri, saç ve göz pigmentasyonunda önemli değişiklikler gözlenir. Sadece göz bulguları da görülebilir.
-
Kanama Eğilimi: Hafif travmalarla bile morarmalar, burun ve diş eti kanamaları sık görülür.
-
Göz Bulguları: Fotofobi, şaşılık, azalmış görme keskinliği, saydam veya farklı renklerde irisler, nistagmus ve kırmızı retinal refleks bulunur.
-
Organ Tutulumu: Özellikle Porto Riko'da görülen ailelerde akciğerde şiddetli fibrozis ve inflamatuvar bağırsak hastalıkları görülebilir.
-
-
Yönetim: Kanama riski nedeniyle hastalara aspirin ve non-steroid anti-inflamatuvar ilaçlardan kaçınmaları önerilir.
Griscelli Sendromu (GS)
Griscelli Sendromu (GS), nadir görülen, otozomal resesif bir hastalıktır. Nörolojik anormallikler veya bağışıklık yetmezliği ile birlikte gümüş gibi saç rengiyle kendini gösterir.
-
GS Tip 1: Gümüşümsü gri saç, açık ten, erken başlayan ağır psikomotor gerilik ve normal bağışıklık sistemi bulunur. MYO5A genindeki mutasyondan kaynaklanır.
-
GS Tip 2: Gümüşümsü gri saç, sık deri ve iç organ enfeksiyonları görülür. Nörolojik sorunlar değişkenlik gösterebilir.
-
GS Tip 3: En hafif formdur. Sadece saç ve deride hipopigmentasyon vardır; sinir sistemi ve bağışıklık sistemi normaldir.
Diğer Albinizm İlişkili Sendromlar
-
Elejalde Sendromu (ES): Gümüşümsü gri saç, bronz ten rengi, santral sinir sisteminde ağır disfonksiyon ve normal bağışıklık sistemi ile karakterizedir.
-
Cross Sendromu (CS): Otozomal resesif geçişli bu hastalıkta albinizm, küçük gözler (mikroftalmi), şeffaf olmayan kornea, spastisite, zeka geriliği ve iskelet anomalileri gibi bulgular görülür.
-
Fenilketonüri (PKU): Otozomal resesif geçişli bir metabolizma bozukluğudur. Fenilalanin metabolize edilemediği için pigmentasyon azalır. Erken dönemde diyetle tedavi edilmezse zeka geriliğine neden olur.
-
Menkes Sendromu (MS): X'e bağlı resesif kalıtılan bir bakır metabolizması bozukluğudur. Erken dönemde hipotermi, beslenme güçlüğü, nörolojik dejenerasyon, depigmente ve anormal saç görünümü (örn. pili torti) görülür. Tanı, saç bulguları ve serum bakır/seruloplazmin seviyelerinin düşüklüğü ile konulur.