- Gösterim: 13430
İnkontinentia pigmenti (İP), Bloch-Sulzberger Sendromu olarak da bilinen nadir, sistemik bir hastalıktır. X'e bağlı dominant olarak kalıtılır ve öncelikle cildi etkiler, ancak dişler, saçlar, gözler ve merkezi sinir sistemi gibi dokularda da sorunlara yol açabilir.
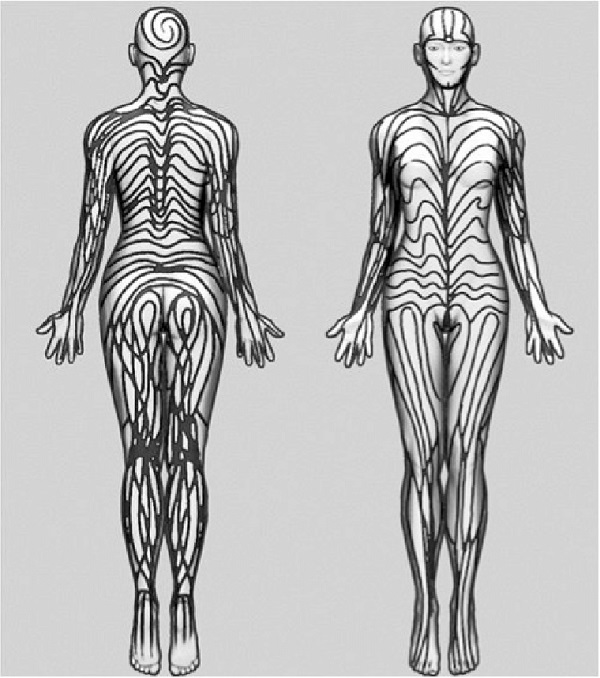
Hastalık, IKBKG genindeki patojenik varyantlardan kaynaklanır. Bu gen, bağışıklık sistemiyle ilgili önemli bir protein olan NEMO'yu kodlar. Bu genetik bozukluk, keratinositlerin etkilendiği ve mutant X kromozomu taşıyan hücrelerin yıkıma uğradığı bir mekanizmayla ortaya çıkar. Bu durum, gövde, kol ve bacaklarda Blaschko çizgilerini takip eden çizgisel veya helezonik, hiperpigmente maküllerle karakterize lezyonlara neden olur.
İP genellikle erkekler için ölümcüldür; bu nedenle hastaların %95'ten fazlası kız çocuklarıdır. Etkilenen erkekler genellikle anne karnında ölür. Hayatta kalan nadir erkek hastalarda ise ya immün yetmezliğe bağlı bir hipomorfik mutasyon, postzigot mutasyon sonucu mozaizm ya da 47,XXY karyotipli Klinefelter sendromu bulunur.
Avrupa'da İP'nin doğum yaygınlığı 100.000'de 1.2 olarak tahmin edilmektedir. Tanı genellikle klinik özelliklere dayanır, ancak IKBKG mutasyonunun tanımlanması, özellikle hafif etkilenen kadınlarda ve doğum öncesi testlerde önemlidir. Deri lezyonları genellikle doğumda klinik tanıyı mümkün kılar.
IP, nöroektodermal dokuları etkileyebilir ve nörolojik bozukluklar, nöbetler veya zihinsel gerilik gibi önemli sakatlıklara yol açabilir. Düşük insidansı ve fenotipik çeşitliliği göz önüne alındığında, vaka serileri hastalığın daha iyi anlaşılması için önemli bir bilgi kaynağıdır.
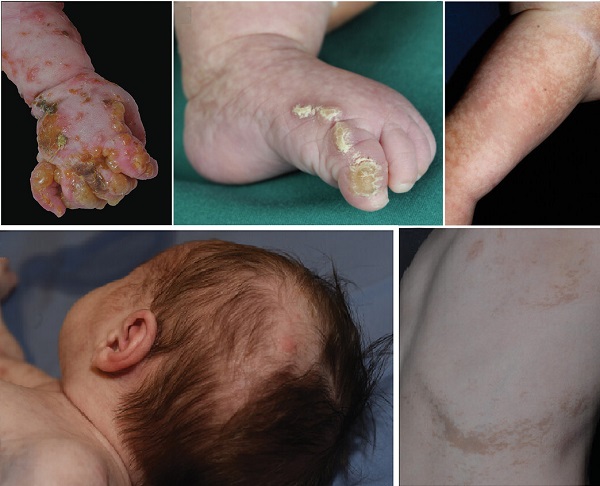
İnkontinentia Pigmenti (İP) Deri Belirtileri ve Dört Evresi
İnkontinentia pigmenti, deri belirtilerini dört farklı evrede gösterir. Ancak bu evrelerin hepsi her hastada görülmeyebilir.
-
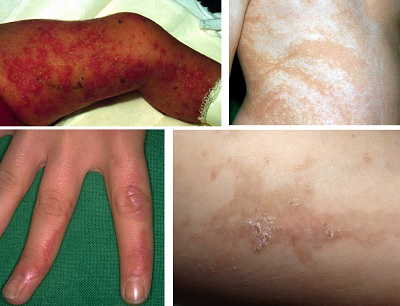
Evre 1 (Vezikülobüllöz Evre): Genellikle doğumla birlikte başlar ve ilk altı hafta içinde ortaya çıkar. Vücudun tamamında görülebilen, en sık kol, bacaklar ve gövdede yerleşen lezyonlar, Blaschko çizgilerini takip eden bir dağılım gösterir. Bu evrede lezyonlarda kızarıklık, veziküller (içi su dolu kabarcıklar) ve püstüller (%91) gözlenir. Hastaların kanında %50'ye varan oranda eozinofili görülebilir. Hastalık kendiliğinden gerileyerek ikinci evreye geçer, ancak bu süreçte güneş gibi dış etkenler nedeniyle belirtiler yeniden alevlenebilir.
-
Evre 2 (Verrüköz Evre): Birinci evrenin lezyonları iyileştikten birkaç hafta veya ay sonra, üzerlerinde hiperkeratozik papüller ve verrüköz (siğil benzeri) plaklar gelişir. Birinci ve ikinci evre lezyonları ortalama 4 ile 7. aylarda geriler.
-
Evre 3 (Hiperpigmentasyon Evresi): Bu evre, sıklıkla 12 ile 26. haftalar arasında ortaya çıkar ve hastalığa adını veren belirgin bulgudur. Vücutta Blaschko çizgilerini takip eden kahverengi veya gri lekeler (%95) oluşur. Bu lezyonlar, genellikle yaşamın ilk haftalarından ergenliğe kadar görülebilir.
-
Evre 4 (Hipopigmentasyon, Atrofi ve Skarlaşma Evresi): Son evre, hipopigmente (daha açık renkli) çizgilerin oluşmasıyla karakterizedir. Lezyonların olduğu bölgelerde, özellikle alt ekstremitelerde, kıl kökleri ve ter bezleri gibi deri ekleri eksiktir. Bu bölgelerdeki deride atrofi ve skarlar bulunur.
İnkontinentia Pigmenti: Deri Dışı Belirtiler ve Genel Seyir
İnkontinentia pigmenti (İP), sadece cilt belirtileriyle sınırlı değildir. Aslında, vakaların en az %50-80'inde deri dışı organ sistemlerinde de tutulum gözlenir.
Evreleme ve Belirsiz Bulgular
İP'nin deri belirtileri dört evrede ortaya çıksa da, her hasta bu sırayı takip etmez. Özellikle üçüncü ve dördüncü evrelerdeki deri bulguları daha belirsiz olabilir ve doğru teşhis konulmasını zorlaştırabilir. Hastalık, bazen doğrudan üçüncü evreden, yani hiperpigmentasyon (koyu lekeler) ile başlayabilir.
Deri Dışı Tutulumlar
İP'ye eşlik eden sistemik belirtiler şunlardır:
-
Dişler: Hastaların yaklaşık %40'ında diş anomalileri görülür.
-
Merkezi Sinir Sistemi (MSS): Vakaların yaklaşık %30'unda MSS tutulumu gözlenir. Bu durum, nöbetler ve zihinsel gerilik gibi önemli sakatlıklara yol açabilir.
-
Gözler: Hastaların %35'inde göz anomalileri mevcuttur.
-
Tırnaklar: Tırnaklarda anormallikler %40 oranında görülür.
-
Diğer: Kas-iskelet sistemi, kulak, kalp-dolaşım sistemi ve bağışıklık sistemiyle ilgili bozukluklar da İP'ye eşlik edebilir.
Bu durum, İP'nin çok yönlü bir hastalık olduğunu ve tanı ile takibin multidisipliner bir yaklaşımla yapılması gerektiğini göstermektedir.
İnkontinentia pigmenti (İP), sadece deri belirtileriyle sınırlı kalmayan, vücudun farklı sistemlerini etkileyen karmaşık bir hastalıktır.
-
Tırnaklar: Tırnaklarda hafif çukurlaşmadan (pitting) onikogrifozise (kuş pençesi tırnak) kadar değişen deformasyonlar görülebilir.
-
Saçlar: Saç dökülmesi (alopesi), saç tellerinde matlık, sertlik ve fırça benzeri bir görünüm bulunabilir.
-
Merkezi Sinir Sistemi: Mental gelişim geriliği, oligofreni (zeka geriliği), tek veya çift taraflı felçler (hemipleji, spastik tetrapleji), küçük kafa çevresi (mikrosefali) ve beyin su toplaması (hidrosefali) gibi ciddi nörolojik belirtiler bildirilmiştir.
-
Gözler: Gözde şaşılık, retinal damar anomalileri, görme kaybı, göz küresinin küçük olması (mikroftalmi), katarakt ve optik sinir atrofisi gibi bulgulara rastlanabilir.
-
Dişler: Diş anomalileri yaşam boyu kalıcı olduğundan tanı koymada önemli rol oynar. Dişlerin eksik olması (hipodonti), gelişimlerinde gecikme ve konik şekilde dişler görülebilir.
-
Diğer Anomaliler: Hastalığa aksesuar meme ucu, meme ucu hipoplazisi, meme hipoplazisi veya aplazisi (gelişim geriliği veya yokluğu) eşlik edebilir. Ayrıca, eklem kontraktürleri, omurga eğriliği (skolyoz) ve kulak anomalileri de görülebilmektedir.