
- Gösterim: 96994
Deri lenfomaları, savunma sisteminin kan hücreleri olan B ve T lenfositlerden köken alan, dermatolojinin önemli hastalıklarından bir grubunu oluşturmaktadır.
Deri lenfomaları ikiye (2) ayrılır:
-
Primer Deri Lenfomaları (PKL): Klinik olarak deriye sınırlı, tanı konduğunda ve altı (6) ay sonrasına kadar deri dışı belirti göstermeyen lenfomalardır.
-
Sekonder Deri Lenfomaları: Lenfomanın deriye yayılması ile ortaya çıkmaktadır.
Primer Kutanöz B Hücreli Lenfoma (PKBHL)
Derinin B lenfosit hücreli lenfomaları ise deri dışı yayılımı bulunmayan, gelişiminin farklı evrelerindeki B hücrelerinden kaynaklanan lenfomaların bir alt grubudur. Derinin primer T hücreli lenfomalarından daha seyrek görülür. Primer deri lenfomalarının yüzde 20-25'ini oluşturur. Çoğu düşük dereceli, yavaş seyirli ve iyi prognozludur. Tanı için en az altı (6) aydır lezyonların sadece deriye sınırlı kalması gerekmektedir.
Derinin B hücreli lenfomaları üçe (3) ayrılmaktadır:
-
Derinin Primer Folikülleri Hedef Alan B Hücreli Lenfoma
-
Derinin Primer Marjinal Zon B Hücreli Lenfoması
-
Derinin Primer Büyük B Hücreli Lenfoması
1. Derinin Primer Folikülleri Hedef Alan B Hücreli Lenfoma
-
Derinin B hücreli lenfomalarının en sık görülen tipidir. Sıklıkla baş ve gövdede yerleşerek yavaş seyir gösterir.
-
Lezyonlar sıklıkla saçlı deri, alın, boyun ve gövdede, nadiren bacaklarda gözlenir. Sırt yerleşimli lezyonlar "Crosti lenfoması" veya "sırtın retikülohistiyositoması" olarak adlandırılmıştır.
-
Hastalık tek veya gruplar oluşturan, kırmızı kahverengi, deriden hafif kabarık plak ve tümörler şeklindedir; bazı lezyonların etrafında yuvarlak tarzda kızarıklık gözlenir. Bunların üzerinde açık yara (ülserasyon) gelişimi nadirdir ve hastaya lezyonlar bir şikâyet vermemektedir.
-
Lenfomanın çok fazla anatomik alanda yerleşimi, hastalığın seyrinin olumsuz olacağı anlamına gelmektedir. Hastalık tedavisiz bırakıldığında lezyonlar genişleyebilir.
-
Deri dışı yayılım nadirdir. Sıklıkla yaşamın 50’li ve 60’lı yıllarında gözlenir. 5 yıllık sağ kalım oranı yüzde 95’ten fazladır. Olguların yüzde 30’unda lezyonlarda tekrarlama görülmektedir ancak bu durum kötü prognoz göstergesi değildir.
-
Tedavi seçenekleri: Radyoterapi, klorambusil, çoklu kemoterapi (CHOP), intralezyonel anti CD20 monoklonal antikorları, çok sayıda lezyon varlığında interferon, IL-2, sistemik steroid uygulaması ve soliter lezyonlarda eksizyon şeklinde sıralanabilir. Tekrarlama eğiliminde olabilen soliter lezyonlara radyoterapi tedavisi eklenebilmektedir.
2. Derinin Primer Marjinal Zon B Hücreli Lenfoması
-
Lenfoma hücreleri deride foliküler merkezin marjinal zonuna yerleşir. Lenfoma hücreleri plazma hücreleri, lenfoplazmositoid hücreler ve marjinal zon B hücreleridir.
-
Sık tekrar etmesine rağmen prognoz çok iyidir.
-
Klinikte tek, nadiren fazla odaklı kırmızı kahverengi, deriden hafif kabarık plak ve nodüller gözlenir. Gövde, kol veya bacaklarda ancak özellikle de kollara yerleşmektedir. Lezyonlarda yara (ülserasyon) nadiren görülür. Deri dışı tutulum nadirdir. Bazı lezyonlarda anetoderma geliştiği bildirilmiştir.
-
Etiyolojide; B. burgdorferi enfeksiyonu, EBV enfeksiyonu, dövme pigmentleri, tekrarlayan HSV-1 enfeksiyonu, influenza, hepatit A aşılaması, AIDS’li hastalar ve immünsüpresan ajan kullanımı gibi antijenik uyarılar suçlanmaktadır.
-
Lezyonlar akrodermatitis kronika atrofikans tarafından tutulmuş alanlar üzerinde de ortaya çıkabilir ve eritema kronikum migrans ile de ilişkilidir.
-
Bu deri lenfomanın 5 yıllık sağ kalım oranı yüzde 100’e yakındır.
-
Tedavi: Düşük doz radyoterapi veya cerrahi eksizyon önceliklidir. Borrelia ile ilişkili olgularda ve erken evrede sistemik antibiyotik verilebilir. İntralezyonel/subkutan IFN alfa, çok sayıda lezyonu olan olgularda oral klorambusil, anti CD20 monoklonal antikoru olan rituksimab verilebilirken sınırlı sayıda lezyonu olan hastalarda altı (6) ayda bir değerlendirilmek üzere takip önerilmektedir.
3A. Derinin Primer Büyük B Hücreli Lenfoması (Bacak Tipi)
-
Karakteristik olarak bacakta yerleşen (yüzde 10-15’i bacak dışı) fakat diğer bölgeleri de tutabilen bir lenfomadır. Baş-boyunda yerleşen büyük B hücreli lenfomalardan çok daha agresif seyirlidir ve deri dışı yayılım sıktır.
-
Sıklıkla yaşlılarda, özellikle kadınlarda görülür. Klinikte kırmızı kahverengi, mavimsi, tek ya da gruplar oluşturan tümör ve plaklar görülür.
-
Deri dışı yayılım sıktır. Ülserasyon sık görülür.
-
Prognoz diğer iki tipe göre daha kötüdür. 5 yıllık sürvi yaklaşık yüzde 50 civarındadır.
-
Tedavi seçenekleri: Çoklu kemoterapi, radyoterapi, rituksimab ile kombinasyon (R-CHOP) bulunmaktadır.
3B. Derinin Primer Büyük B Hücreli Lenfoması (Diğer)
-
Sıklıkla baş, gövde ve bacaklarda yerleşir. Primer kutanöz T hücre/histiyosit zengin B hücreli lenfomalar ve primer kutanöz intravasküler büyük B hücreli lenfomalar bu gruptadır.
İntravasküler Büyük B Hücreli Lenfoma
-
Büyük B hücreli lenfomaların iyi tanımlanmış bir alt tipidir. Malign anjioendotelyoma olarak da bilinir.
-
Merkezi sinir sistemi, böbrekler, akciğer, karaciğer ve deri tutulumu sıktır. Prognozu kötüdür.
-
Kliniği pannikülit veya sklerotik bağ doku hastalığına benzeyebilir. Sıklıkla bacakların alt kısmı veya gövdede kırmızı-mavi infiltre plak veya yama şeklinde gözlenir. Papül ve nodüller de olabilir ve ülserasyon sıktır. Telenjiyektatik deri lezyonları da eşlik edebilir.
-
Hastalar sıklıkla ateşli, tekrarlayan enfeksiyon bulguları olan kronik hastalardır. Merkezi sinir sistemi ve akciğer tutulumu sık, lenf nodu tutulumu nadirdir.
-
Tedavide: Çoklu kemoterapi uygulanabilmektedir. Prognoz kötüdür. Merkezi sinir sistemi ve akciğer tutulumu olanlarda yaşam süreleri aylarla sınırlıdır. 3 yıllık yaşam süresi yaklaşık yüzde 20’dir.
Primer Kutanöz T Hücreli Lenfoma (PKTHL)
Primer kutanöz lenfomaların yüzde 65'ini (65%) T hücreli lenfomalar oluşturur.
Mikozis Fungoides (MF)
Mikozis Fungoides (MF), en yaygın görülen primer deri lenfomasıdır ve PKTHL'lerin yüzde 40’ını oluşturur.
-
Görülme sıklığı: 0,3-1,0/100.000 kişidir.
-
Hastaların çoğu erişkindir ve ortalama 40 yaş civarında başlar.
-
Erkekler daha fazla etkilenir (Erkek/Kadın oranı 1,6-2'dir).
-
Klinik olarak yama, klasik ve atipik deri belirtileri gösteren farklı klinik formlarda görülebilir.
MF'in Klinik Tipleri ve Evreleri
1. Yama Evresi
-
Erken yama evresinde vücutta tek veya çok sayıda, kırmızı ve üzeri kepekli döküntüler izlenir. Boyutları değişmekle birlikte sınırları belirgindir. Rengi turuncudan morumsu-kırmızıya kadar değişebilir.
-
Hafif kaşıntılı veya kaşıntısız olabilir. Zaman zaman kendiliğinden kaybolup, yeniden çıkabilir.
-
Yama döneminde birçok cilt hastalığına benzediği için MF’in klinik tanısı oldukça güçtür. Biyopsi ve şüphelenilen hastalarda biyopsilerin tekrarlanması gerekmektedir.
-
Genellikle vücudun kapalı yerlerinde yerleşir.
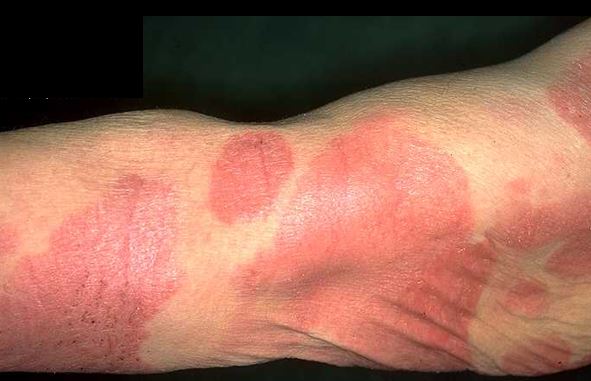
2. Plak Evresi
-
Plak evresinin deri belirtileri yama döneminden aylar veya yıllar sonra başlayabileceği gibi, doğrudan plaklar ile de başlayabilir.
-
Plaklar keskin sınırlı, deriden hafif kabarık, kırmızıdan mor, kahverengine değişen renkte, deride yayılmalar göstermektedir.
-
Dağılım yine yama evresinde olduğu gibidir ve genellikle vücudun kapalı yerlerinde yerleşir. Hastaların büyük bir çoğunluğunda şiddetli kaşıntı vardır.
-
Plakların büyüklüğü ve kalınlığı yavaşça artış gösterir, bu plakların ortasında yer yer keskin sınırlı normal deri adaları bulunabilir. Kendiliğinden iyileşmeler olabilir ancak yama evresi kadar sık gözlenmez.
-
Plakların şekilleri yuvarlak, iç içe geçmiş yuvarlaklar ya da at nalı şeklini alabilirler. Nadiren ikincil enfeksiyonun plaklara yerleşmesi, plaklarda deri bütünlüğünde bozulma ve yaralar yapabilmektedir.
-
Bu dönemde lenf bezlerinde büyüme (lenfadenopati) bulunabilir. Biyopsi en doğru tanısal yöntemdir.
-
Son yıllarda plakların kalınlığının hastalığın klinik şiddetini belirlediği gösterilmiştir.
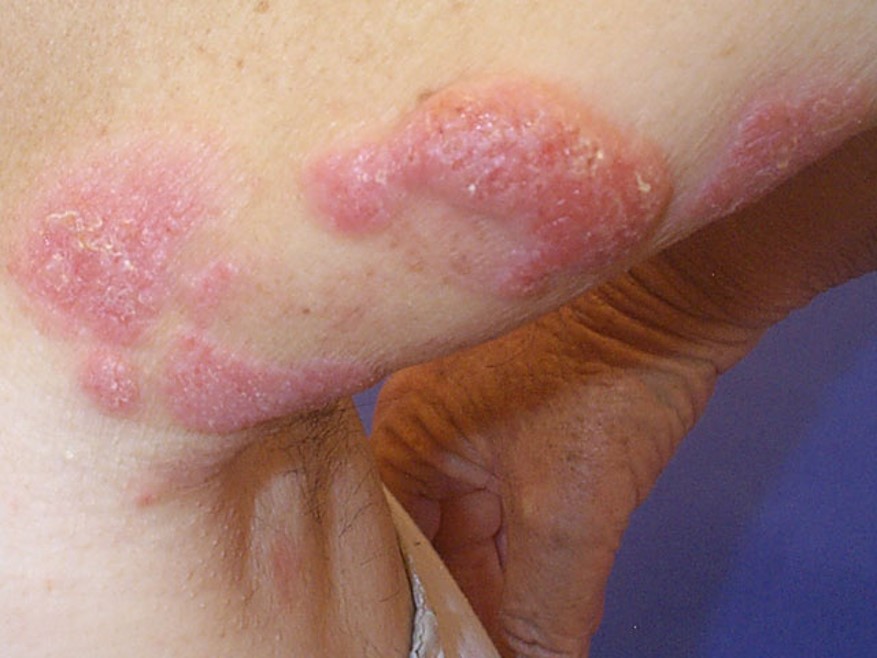
3. Tümör Evresi
-
Plaklar üzerinde gelişebildiği gibi doğrudan tümör olarak da başlayabilir. Doğrudan başlayan tipinde erken evre belirtilerinden herhangi biri olmaksızın hızla büyüyen büyük tümöral oluşumlar vardır ve buna "tumor de emblee" denilmektedir.
-
Tümöral evre lezyonları herhangi bir vücut bölgesinde bulunmasına rağmen en sık yüz ve kıvrım bölgelerinde yerleşir.
-
Nodüller kırmızı kahve veya morumsu kırmızı renkte, düz yüzeylidir ama genellikle ülserleşir ve sekonder olarak enfeksiyon gelişir. Değişik büyüklükte olabilirler.
-
Çoğu hastada sistemik belirtiler eşlik eder. Çok nadir de olsa tedavisiz gerileme görülebilir. Bu dönemde hastalığın genel seyri diğer evrelerden daha kötüdür.
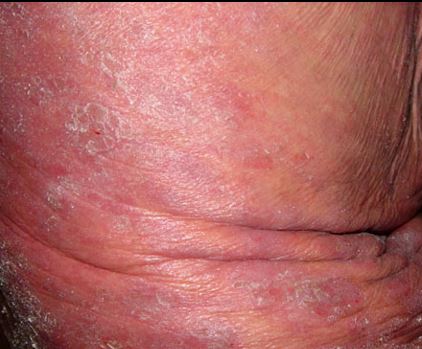
4. Eritrodermi Evresi
-
Doğrudan başlayabileceği gibi MF’in diğer lezyonlarından da gelişebilir. Nadir görülür.
-
Vücudun yüzde 80’inden fazlası parlak kırmızı renkte ve kepeklerle kaplıdır; karakteristik olarak simetrik sağlam deri adacıkları bulunur.
-
Yüz tutulumu "aslan yüzü" görünümüne neden olur.
-
Ateş, titreme, iştahsızlık, kilo kaybı ve çok şiddetli kaşıntı vardır. El-içi, ayak-tabanında da kızarıklık (eritem), kepeklenme (skuam) ve çatlaklar (fissürler) bulunabilir.
-
Lenfadenopati genellikle vardır. Alopesi, ektropion, tırnak distrofisi olabilir.
-
Bu dönemin histolojisi oldukça değişkendir. Eritrodermiye neden olan diğer dermatozlar ile ayırıcı tanısı yapılmalıdır.
Sezary Sendromu (SS)
-
Tarihsel olarak eritrodermi, periferal lenfadenopati (LAP) ve periferik kanda yüzde 5’ten daha fazla atipik mononükleer hücre (Sezary hücresi) bulunması olarak tanımlanmaktadır.
-
Tüm PKTHL’ların yüzde 5’inden daha azını oluşturur, hemen hemen tümü erişkinlerde görülür.
-
Belirgin olarak vücutta deri dökülmesi, ödem ve likenifikasyonun eşlik ettiği, şiddetli kaşıntılı eritrodermi ile karakterizedir.
-
Yaygın bulgular: Lenfadenopati, alopesi, göz kapaklarının dışa dönmesi (ektropion), tırnak düzensizlikleri ve el içi ile ayak tabanında sertleşme ve derin çatlaklar, karaciğer ve dalakta büyüme.
-
Hastalığın klinik seyri kötü, tanıdan itibaren ortalama yaşam süresi 2-4 yıl olarak bildirilmektedir. 5 yıllık yaşam şansı yüzde 10-20 arasındadır.
-
Ölüm nedeni genellikle şiddetli fırsatçı enfeksiyonlar veya hastalığın daha agresif seyrettiği "anaplastik büyük hücreli lenfomaya" dönüşmesidir.
Yeni SS Tanı Kriterleri:
-
Total lenfosit sayısının yüzde 20’sinden fazlasının veya yaklaşık 1000 Sezary hücresinin bulunması.
-
T lenfositlerden CD4/CD8 oranının 10’dan fazla olması ve/veya pan T-hücre antijenleri (CD2, CD3, CD4, CD5)’nden biri veya hepsinin kaybının akım sitometrisi ile gösterilmesi.
-
Artmış lenfosit sayısı ile birlikte moleküler veya sitogenetik metotlar ile periferik kanda T-hücre klonunun gösterilmesi.
-
Kromozomal olarak anormal bir T-hücre klonu.
MF’in Klinik Varyantları
Klinik davranışları klasik MF’e benzediği için farklı olarak kabul edilmemektedir. Hastalığın bu varyantlarına sahip hastalarda sıklıkla vücutlarının diğer alanlarında aynı zamanda klasik mikozis fungoides özelliklerini taşıyan lezyonlar bulunur.
Vezikülo-büllöz MF
-
Yaşlılarda (ortalama 60 yaş) görülür ve cinsiyet farkı yoktur.
-
Klinik olarak normal ya da kırmızı deride çabuk yırtılan vezikül veya büller şeklinde su toplamaları veya tipik MF plakları veya tümöral lezyonları üzerinde görülebilir. Bunlar deride açık yaralara (nekroza) dönebilir.
-
Gövde, kol ve bacaklar en çok bulunan vücut bölgeleridir.
-
MF’de büllöz lezyonlar kötü prognoz belirtisidir, çünkü hastaların yüzde 50’si bir yıl içinde kaybedilmektedir.
Syringotropik/Adneksotropik MF
-
Kutanöz T hücreli lenfoma, ter bezlere yayılan ve tutan çok sayıda küçük deriden kabarmalar (papüllerle) karakterizedir.
-
Klinik olarak kırmızı-kahverengi yamalar, hafifçe kepekli plaklar veya küçük kırmızı veya deri renginde papüller olarak görülür.
-
Tutulan alanlarda kıl kaybı sıklıkla bulunur, terleme yokluğu vakaların üçte birinde vardır. Ülserasyon son derece nadirdir. Günümüze kadar yalnız erkek olgular bildirilmiştir.
Hipopigmente MF
-
Sıklıkla koyu deri renkli Hintli veya Afrika-Amerika orijinli gençlerde görülmesine rağmen zaman zaman açık deri rengine sahip kişilerde de görülebilir.
-
Hastalar genellikle asemptomatik veya hafif kaşıntılı düzensiz kenarlı kepekli hipopigmente yamalar ile başvurmaktadır.
-
Özellikle kasık ve gövde tutulmaktadır.
-
Hipopigmentasyonun nedeni henüz tam olarak bilinmemektedir.
Hiperpigmente MF
-
MF’in bu tipi tek klinik özellik olarak yaygın (diffüz), leke şeklinde (maküler) hiperpigmentasyon (Ashy dermatozuna benzer) ile karakterizedir.
Poikilodermik MF
-
Bu klinik tabloda lezyonlar yaygın veya az sayıda belli bir vücut alanında açık-koyu renk değişimi (hipohiperpigmentasyon), kuruluk, deride incelme ve kılcal damar artışı ile bir arada bulunmaktadır. Bu klinik tablo birlikteliğine poikiloderma denilmektedir.
-
Bu lezyonlar sıklıkla daha önceki yama alanlarında, genellikle de giysilerin kronik olarak sürtünme bölgelerinde ortaya çıkar.
-
Genellikle gövde tutulur, göğüsler, kasık ve kalça alanları da etkilenebilir.
Palmoplantar MF
-
MF’in klasik tipinde el içi-ayak tabanı tutulumu vakaların yüzde 11,5’inde görülür. Ancak vücutta başka bir alanda olmaksızın sadece el içi-ayak tabanından başlaması veya öncelikle etkilenmesi palmoplantar MF olarak adlandırılır ve yüzde 0,6 sıklık oranında görülmektedir.
-
Klinik olarak yuvarlak, hiperpigmente yama ve plaklar, hiperkeratotik lezyonlar, küçük su toplamaları, püstüller, siğillere benzer yapısal değişiklikler, sedefe benzer plaklar, açık yaralar ve tırnak deformiteleri şeklinde olabilir.
-
Klinik seyir genellikle iyidir, çoğu vakada başlangıç alanına sınırlı kalır, nadiren vücuda yayılabilir. Deri dışı sistemik tutulum bildirilmemiştir.
Hiperkeratotik/Verrüköz MF
-
Hiperkeratotik ve siğil benzeri (verrüköz) plaklar olarak bacaklar, yüz ve gövdede görülebilir. Klasik MF lezyonları veya el içi-ayak tabanı tutulumu eşlik edebilir.
Vegetatif/Papillomatöz MF
-
Bu lezyonlar koltuk altı, kasık gibi katlantı alanları, boyun, göğüsler de ortaya çıkar. Dağılım, büyüklük ve rengine bağlı olarak seboreik keratoz veya akantozis nigrikansa benzer.
Pigmente Purpura Benzeri MF
-
Uzun süreli deride küçük kanamalar (purpura) ve pigmentasyon değişimleri ile karakterizedir. MF’in bu tipini pigmente purpurik dermatozdan ayırt edebilmek için yakın takip gerekmektedir.
Püstüler MF
-
Püstüler döküntüler el içi ve ayak tabanı alana sınırlı veya tüm vücutta olabilir.
İktiyoziform MF
-
Nadir görülen klinik bir alt tipidir. Yaygın iktiyoziform lezyonlara sıklıkla komedona benzer lezyonlar ve/veya foliküler keratotik papüller eşlik eder. Özellikle kol ve bacaklar olmak üzere tüm vücut yüzeyi tutulabilir.
-
Şiddetli kaşıntıya bağlı olarak deride tırnak izleri yaygındır.
-
MF’in bu tipinde oral retinoidlerin PUVA veya UVA tedavisi ile kombinasyonunun en etkili tedavi olduğu gösterilmiştir.
Ayrı Değerlendirilmesi Gereken Klinik Formlar
Folliküler Musinozis
-
Follikülotropik MF, pilotropik MF, follikülosentrik MF olarak da tanımlanmaktadır.
-
Belirgin olarak erkeklerde kadınlardan 4-5 kat fazla görülmektedir.
-
Klinik olarak akneye benzeyen lezyonlar, komedon benzeri tıkaç formasyonu, epidermal kistler, kıl folliküllerine uyan küçük deriden kabarmalar ve hiperkeratozlar, kırmızı yama ve plaklar ve etkilenen alanlarda kıl kaybı gözlenmektedir.
-
Yüz, gövde ve üst gövde en sık etkilenen alanlardır. Şiddetli kaşıntı eşlik eder.
-
Foliküler MF’in klasik MF’e göre daha agresif davrandığı ve prognozunun daha kötü olduğu bilinmektedir. Tümör ve eritrodermi gelişimi daha sıktır.
-
İdiyopatik foliküler müsinozisden ayırt edilmelidir.
Pagetoid Retikülozis
-
Neoplastik T hücrelerinin deride epidermiste çoğalması ile lokalize yama ve plaklar gelişmektedir.
-
Tipik lezyonlar, vücudun yüzde 5’inden daha azını tutan tek, belirgin keskin sınırlı, lokalize kepekli psöriazise benzer veya hiperkeratotik plaklar şeklindedir.
-
Genellikle kol ve bacakların alt kısımlarına yerleşir, yavaş büyüme gösterir ve deri dışı sistemik tutulum yoktur. Prognoz oldukça iyidir.
Granülomatöz Gevşek Deri Sendromu
-
PKTHL’ların oldukça nadir görülen bir tipidir ve deri kıvrımlarında artış veya gevşek deri gelişimi ile karakterizedir.
-
En sık koltuk altı ve kasıkta görülür. Bildirilen vakaların üçte birinde Hodgkin lenfoma ve MF ile birliktelik rapor edilmiştir.
-
Hastalarda poikiloderma ile büyük infiltre alanlar gelişir, atrofik hal alır ve atrofik deri kıvrımlarında elastik liflerin kaybı ile sonuçlanır. Deride yer yer gevşeklik ve sarkıklıklar, yer yer fibrotik bantlar vardır.
-
Hastaların çoğu iyi seyir gösterir. Orta yaşlı erişkin kadınlarda daha sık görülür.
CD30+ Lenfoproliferatif Hastalıklar
İkinci en sık PKTHL formunu oluştururlar. Bu grupta lenfomatoid papülozis, primer kutanöz anaplastik büyük hücreli lenfoma (PKABHL) ve borderline olgular yer alır.
Lenfomatoid Papülozis
-
Kronik seyirli olup, aynı anda papüler, papülonekrotik ve nodüler lezyonlar bir arada bulunabilir.
-
Lezyonlar kendiliğinden skar bırakarak iyileşip nüksedebilir.
-
Klinik tablo oldukça iyi huylu (benign) görünmesine rağmen histolojik olarak büyük hücreli anaplastik lenfoma, MF veya Hodgkin hastalığına benzeyen üç (3) farklı tipi bulunur.
-
Hastaların yüzde 10-20’si malign lenfomaya dönüşebilir.
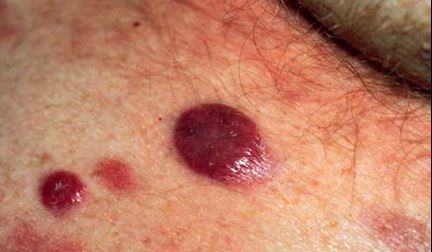
Primer Kutanöz Anaplastik Büyük Hücreli Lenfoma (PKABHL)
-
Daha çok erişkinlerde görülür, MF’e göre daha lokalizedir.
-
Çoğu hastada tek veya çok sayıda ülsere olabilen kırmızı kahverengi nodüller bulunur.
-
Bölgesel lenf nodu tutulumu olsa bile 5 yıllık yaşam şansı yüzde 95’tir.
-
Cerrahi ve radyoterapi tek lezyonlar için ilk seçenektir. Düşük doz metotreksat gruplu veya multiple lezyonlar için etkilidir.
Subkutan Pannikülit Benzeri PKTHL
-
Kutanöz lenfomaların yüzde 1’ini oluşturur.
-
Bacaklarda, kollarda, gövdede, nadiren yüzde lokalize, deri altı derin yerleşimli kırmızı mor renkli tümöral nodüller ile karakterizedir.
-
Nadiren spontan gerileyebilir veya genç hastalarda tekrar edebilir.
-
Sistemik semptomlar eşlik edebilir.
-
Prognoz iyi, 5 yıllık yaşam şansı yüzde 80’dir.