
- Gösterim: 6458
Bazı klasik otozomal dominant geçişli deri hastalıkları mozaisizm nedeniyle sadece Blaschko çizgilerini izleyen lezyonlar şeklinde karşımıza çıkabilmektedir. Bu durumda bu bulguların tüm vücutta yaygın görülmesi durumunda hangi hastalığa işaret edeceğini bilmek doğru tanıya ulaşmak ve aileye genetik danışmanlık vermek adına önemlidir. Ancak Blaschko çizgilerini izleyen lezyonlar karşımıza çıkan bulgular her zaman jeneralize bir hastalıkla eşleşmeyebilir. Bu durum aşağıda anlatılan otozomal dominant ölümcül hastalıklarda yani sadece mozaik durumlarda hayatla bağdaşan durumlarda görülür. Ölümcül olmayan OD letal olmayan bir genodermatozun mozaik deri lezyonu şeklinde belirti vermesine iki şekilde rastlanabilir.
A. Tip1 segmental bulgu
Embriyonik gelişim sırasında bir genin tek bir alelinde otozomal dominant hastalığa ait birden ortaya çıkan mutasyon olması durumudur. Bu durumda bu hastalığın bulguları mutant hücrelerin dağılımına karşılık gelen bölgelerde Blaschko çizgileri izleyen veya diğer mozaik deri lezyonları şeklinde ortaya çıkar. Kalan deri bölgeleri ve bu bölgelerdeki hücrelerin genotipi normaldir. Blaschko çizgileri tipinde görülen bulgular tüm vücutta yaygın hastalığın bulgularının morfolojik olarak aynısıdır. Çoğunlukla bu durumda aile öyküsü yoktur. Hastada mutasyon gonadları etkilemişse sonraki nesillere hastalığı aktarma riski vardır (yaygın lezyon ve genital bölgelezyonları ile seyrediyorsa).
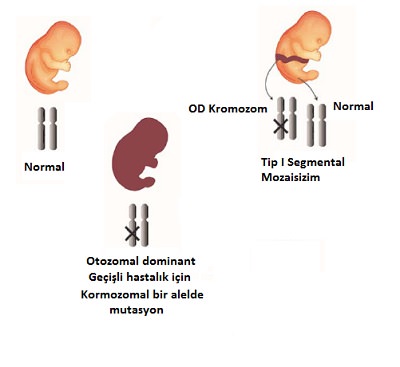
A.I. Sporadik lineer epidermal nevüs (iktiyozis histriks, epidermolitik verrükoz epidermal nevüs, lineer büllöz iktiyoziform eritroderma)
Bu durumda jeneralize bir hastalık olan büllöz konjenital iktiyoziform eritroderma (KİE) mozaik olarak epidermal nevüs şeklinde karşımıza çıkmakta ve histopatolojisi incelendiğinde epidermolitik hiperkeratoz görülmektedir. Sıklıkla doğumda var olan Blaschko çizgilerini izleyen kahverengi-ten renginde siğil şeklinde lezyonlarla karakterizedir. Bu hastaların gonadlarında da mutasyon mevcutsa çocuklarına bu hastalığı geçirme riski vardır.
A.II. Unilateral Palmoplantar Verrüköz Nevüs
Keratin 16 gen mutasyonunu mozaik şeklinde bulunduran bireylerde tek taraflı ve Blaschko çizgilerini izleyen verrüköz nevüs bulgularına rastlanmaktadır. Bu durumun pakionişi konjenita tip 1 ‘in tip 1 segmental bulgusu olduğu düşünülmektedir.
A.III. Diğer non-epidermolitik lineer verrüköz nevüsler
Bazı verrüköz epidermal nevüslerin akantozis nigrikansın lokalize formları olduğu düşünülmektedir. Crouzon kraniosinostozu, akantozis nigrikans ile ilişkili bir sendrom olup FGFR3 (fibroblast büyüme faktörü reseptörü) genindeki mutasyona bağlı olarak oluştuğundan FGFR genlerinin bu lokalize nevüs formlarıyla ilişkili olduğu düşünülmektedir.
A.IV. Nevüs Komedonikus (Akneiform Nevüs )
Apert sendromu otozomal dominant geçişli bir hastalık olup FGFR2 gen mutasyonuna bağlı oluşmaktadır. Kraniyosinostoz, klinodaktili, sindaktili ve şiddetli akne ile karakterizedir. Bu sendromun mozaik durumlarında nevus komedonikus görülmektedir. Nevüs komedonikus genellikle doğumdan itibaren vardır ancak ergenlikle beraber daha belirgin hale gelmektedir. Keratin dolu çukurların lineer çizildiği bazen verrüköz tabanlı lezyonlardır.
A.V. Nevüs Kornikulatus
Nevüs kornikulatus yeni bir akantolitik hastalık olarak tanımlanmış olup Blaschko çizgileri dağılımda hiperkeratotik plağa eşlik eden filiform hiperkeratoz, boynuz benzeri çıkıntılar ve dev komedonlar lezyonlarla karakterizedir.
A.VI. Lineer Darier Hastalığı
Lineer Darier hastalığının morfolojik bulguları jeneralize form ile aynı olup ATP2A2 gen mutasyonuna bağlı olarak ortaya çıkmaktadır.
A.VII. Lineer Hailey Hailey Hastalığı ve Tekrarlayan Lineer Akantolitik Dermatoz
Hailey Hailey hastalığının lineer lezyonlar şeklinde seyretmesi tip 1 segmental bulgu olarak öne sürülmüş ve hastalığa neden olan mutasyonun postzigotik dönemde mozaik olarak görülmesine bağlanmıştır.
A.VIII. Lineer Porokeratoz
Porokeratoz kenarları hiperkeratotik halka ile çevrili merkezi atrofik plaklarla karakterize genetik bir hastalık grubudur. Altı adet alt varyant bildirilmiş olup hepsinin histopatolojisinde kornoid lamella gözlenmektedir. “Klasik porokeratozis Mibelli”’de aile hikayesinin sık olması OD bir hastalığı düşündürmektedir. “Dissemine aktinik süperfisyal porokeratoz (DSAP)” OD geçişli olup UV ışınlarına temas sonrası güneşe maruz kalan bölgelerde porokeratoz lezyonlarıyla karakterizedir. “Lineer porokeratoz” çoğunlukla çocuklukta başlar ve Blaschko çizgilerine dağılımlı lezyonlarla seyreder. DSAP’ nin segmental dağılımlı lezyonlar şeklinde görülmesi DSAP’ye neden olan mutasyonun postzigotik dönemde oluşmasına (Tip 1 segmental bulgu) bağlanmıştır. Bununla uyumlu olarak çocukluğunda lineer porokeratozu olan bir kişide erişkin yaşta DSAP lezyonlarının görülmesi heterozigositenin kaybı olarak yorumlanmıştır. Bu durumdapostzigotik dönemde oluşmasına (Tip 1 segmental bulgu) bağlanmıştır. Bununla uyumlu olarak çocukluğunda lineer porokeratozu olan bir kişide erişkin yaşta DSAP lezyonlarının görülmesi heterozigositenin kaybı olarak yorumlanmıştır. Bu durumda DSAP için heterozigot mutasyonu taşıyan embriyoda aynı genin ikinci alelinde mutasyon oluşmasıyla tip 2 segmental bulgular ortaya çıkmaktadır. Bu bireyde lezyonlar erken yaşta daha şiddetli olarak Blaschko çizgilerini izleyen dağılımda görülmektedir. Dikkat çekici olarak bu hastanın “lineer porokeratoz” kliniği zamanlama olarak daha geç oluşan bir mutasyona bağlı olmasına rağmen DSAP kliniği lineer porokeratoz lezyonlarından daha sonra ortaya çıkmaktadır. Lineer dağılımlı lezyonlarda malign potansiyelin daha fazla oluşu heterozigositenin kaybına bağlanmıştır.
A.IX. Lineer Bazal Hücreli Karsinom
Nevoid bazal hücreli karsinom (NBCC) sendromu (Gorlin sendromu) multipl BCC, geniş yüz, çene kemiği kistleri, palmar pits ve kaburga anomalileri ile karakterizedir. Bu hastalık 9. kromozomdaki PTCH gen mutasyonuna sonucu oluşmakta ve tümörler heterozigositenin kaybolmasıyla ortaya çıkmaktadır. Lineer yerleşimli bazal hücreli nevüs ya da BCC izole bir bulgu olarak ya da NBCC sendromun bir parçası olarak görülebilir. Normal bir bireyde lineer yerleşimli BCC lezyonlarının görülmesinin NBCC sendromunun mozaik formu olduğunu (Tip 1 segmental) öne süren yazarlar olmakla birlikte nonsendromik herediter multipl BCC’nin heterozigosite kaybıyla ortaya çıkan tip 2 segmental bulgu olduğu düşünülmektedir.
A.X. Segmental Nörofibromatozis Tip 1
Bazı hastalarda nörofibromlar ve cafe au-lait makülleri gibi NF-1 lezyonlarının segmental yerleştiği görülmektedir. Bu durum 17. kromozomdaki NF-1 geninde postzigotik mutasyon sonucu oluşan mozaisizme bağlanmıştır. Bu hastalarda pigmenter lezyonların Blaschko çizgilerine yerleştiği ancak nörofibromların dermatomal tipde görüldüğü bildirilmiştir.
A.XI. Lineer Anjiofibroma
Anjiofibromların segmental dizilimli lezyonlar şeklinde görülmesinin OD kalıtımlı bir hastalık olan tüberoskleroza neden olan genler açısından mozaisizmi yansıttığı düşünülmektedir.
A.XII. Unilateral Nevoid telenjiektazi
Unilateral nevoid telenjiektazi dermal vasküler bir malformasyon olup ilk kez Blaschko tarafından tanımlanmıştır. Sanıldığından daha sık rastlanan ancak raporlanmayan ya da farkedilmeyen bir durum olduğu düşünülmektedir. Genellikle sonradan olmakla birlikte konjenital olarak da görülebilir. Konjenital formlar otozomal dominant geçişlidir. Nedeni tam olarak bilinmemekte ancak hiperöstrojenik durumlar ve vazoaktif diğer maddeler suçlanmaktadır. Ancak erkeklerde de görülmesi hiperöstrojenik durumlar dışında da faktörlerin rol aldığını göstermektedir. Altta yatan somatik mozaisizmin çevresel faktörlerin de etkisiyle bu durumu ortaya çıkardığı düşünülmektedir. Daha çok oral kontraseptif kullanan, gebe ya da kronik karaciğer hastalığı bulunan kadınlarda ergenlik döneminde başlar.
Çeşitli sayıda telenjiektazilerin lineer, dermatomal ya da Blaschko çizgileri üzerinde dağıldığı görülür. Unilateral nevoid telenjiektazinin herediter benign telenjiektazinin mozaik formu (OD hastalığın tip 1 segmental bulgusu) olduğunu öne süren yazarlar olmakla beraber bazı yazarlar unilateral nevoid telenjiektaziyi paradominant mekanizmayla gelişen ve genetik olarak aktarılan hastalıklar arasında sınıflamaktadır.
A.XIII. Diğer Lineer Benign Adneksiyel Tümörler
Siringomlar, ekrin spiradenomlar, silindromlar ve trikoepitelyomlar gibi derinin adneksiyel tümörlerinin nadir vakalarda lineer dizilimde görüldüğü bildirilmiştir. Bu tümörlerin lineer lezyonlar şeklinde görülmesinin mozaisizme bağlı olabileceği düşünülmektedir.
A.XIV. Bazaloid folliküler hamartom
Bazaloid folliküler hamartom derinin nadir bir adneksiyel tümörü olup kalıtımsal (herediter) veya kalıtımsal olmayan (nonherediter) formlarda görülebilir. Lineer unilateral bazaloid folliküler hamartom (LUBFH) nonherediter olup henüz tanımlanmamış bir genin postzigotik mutasyonu sonucu oluştuğu düşünülmektedir. Söz konusu genin bulunduğu mozaik bölgelerde lezyonların Blaschko çizgileri dağılımıyla karakterizedir. Lezyonlar folliküler tıkaç, fibröz değişiklikler, hipo/hiperpigmentasyon ve adneks kaybının eşlik ettiği papül ve plaklarla seyreder.
B. Tip 2 segmental bulgu
Bu durumda şiddetli mozaik lezyonlar hafif veya klinik olarak belirgin olmayan jeneralize bir hastalık üzerinde görülür. Bu hastalarda bir genin tek bir alelindeki (heterozigot) mutasyon sonucu OD bir hastalık bulunmakta ve postzigotik dönemde aynı genin diğer alelinde de mutasyon gerçekleşmesiyle heterozigozitenin yitirilmesi (LOH) olarak adlandırılan durum oluşmaktadır. Burada lezyonlar ikinci mutasyonu da taşıyan hücrelere karşılık gelen bölgelerde Blaschko çizgileri tipte daha şiddetli ve/veya daha erken belirmektedir . Bu hastalarda aile öyküsü olabilir veya olmayabilir. Bunun nedeni söz konusu bireydeki heterozigot mutasyon ebeveynlerin birinden kalıtılmış olabilir veya de novo ortaya çıkmış olabilir.
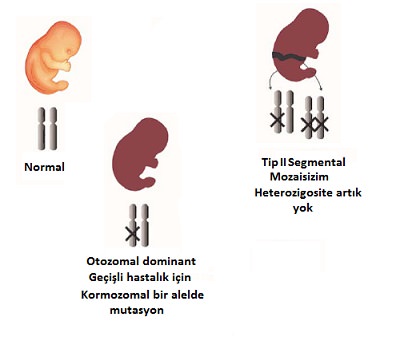
ip 2 segmental bulgusu olan birey heterozigot mutasyon taşıdığından (lineer lezyon bölgesindeki hücreler hariç) çocuklarına hastalığı geçirme riski %50 dir. Tip 2 segmental bulgular moleküler düzeyde yapılan çalışmalarla Hailey Hailey Hastalığı, Neurofibromatozis tip 1, Legius sendromu (Nörofibromatozis tip 1 benzeri sendrom: cafe-au-lait maküller, aksiller çillenme ve makrosefali ile karakterize, SPRED1 gen mutasyonu), SOLAMEN sendromu (PTEN geni mutasyonuna bağlı, Cowden hastalığının klasik bulguları ile Proteus sendromuna benzer bulgular- segmental devlik, arteriovenöz malformasyonlar, lenfatik vasküler malformasyonlar, lipomatozis, lineer epidermal nevüs), Darier hastalığı ve ailevi glomuvenöz malformasyonlarda tanımlanmıştır.
Yine tip 2 segmental bulguyu yansıttığı düşünülen akantozis nigrikans, epidermolitik hiperkeratoz vakaları da bildirilmiştir.
Mozaisizm ile Kurtarılan Otozomal Dominant Letal Deri Hastalıkları
Otozomal genlerde görülen bazı mutasyonlar vücudun tüm hücrelerinde görüldüğünde erken embriyonik ölümle sonuçlanmaktadır. Dolayısıyla bu tip mutasyonların neden olduğu nihai fenotip sadece mozaik durumlarda karşımıza çıkmakta ve jeneralize hastalık olarak hiçbir zaman görülmemektedir. Bu durumların letal OD hastalıklarla veya otozomal resesif letal bir hastalık taşıyan bireyde heterozigositenin kaybıyla (LOH) oluştuğu düşünülmektedir.


