
- Gösterim: 10602
Blaschko çizgilerini takip eden birçok cilt hastalığı, X kromozomuna bağlı olarak ortaya çıkmaktadır. Bu lezyonlara kadınlarda daha sık rastlanılması, tüm kadınlarda görülen fonksiyonel X kromozomu mozaisizmine bağlanmıştır. X'e bağlı bir hastalık kadınlarda görüldüğünde, fonksiyonel mozaisizm (liyonizasyon) nedeniyle hastalığa ait fenotip, mutasyon taşıyan X kromozomunun aktif olduğu dokularda ortaya çıkabilmektedir.
Dolayısıyla, bu kadınlarda mozaisizme bağlı deri bulguları görülebilir ve bunlar genellikle Blaschko çizgilerine paraleldir. İnkontinensia pigmenti'de olduğu gibi, mozaisizmin liyonizasyonu embriyogenezin erken döneminde gerçekleştiği için, X'e bağlı hastalıklardaki çizgisel lezyonlar genellikle çok sayıda ve dardır. Ancak, X'e bağlı mozaisizmi bulunan hastalarda deri lezyonlarının her zaman Blaschko çizgileri üzerine yerleşmediği unutulmamalıdır. Çünkü mozaisizm, bu hastalıklarda liyonizasyon dışında X kromozomuna ait somatik mutasyona da bağlı olabilmektedir. Bu duruma örnek olarak, X'e bağlı konjenital jeneralize hipertrikozda blok şekilli ya da CHILD sendromunda lateralizasyon şeklindeki mozaisizm lezyonları verilebilir.
Yine X'e bağlı bazı hastalıklarda, etkilenen hücreler derinin kalıcı elemanları olmadığında (dolaşımdaki lökositler, endotel hücreleri gibi), hastalığı taşıyan kadınlarda liyonizasyona bağlı mozaisizm bulunsa bile, klinikte görülebilir bir çizgisel tip olmayabilir (kronik granülomatöz hastalık, Fabry hastalığı - alfa galaktosidaz eksikliği, Wiskott-Aldrich sendromu gibi).
Steroid sülfataz eksikliği taşıyıcısı (X'e bağlı resesif iktiyoz) olan kadınlarda beklenen lineer iktiyoz lezyonlarının görülmeyişi ise bu genin kısmi olarak aktif olmadığını düşündürmektedir. Steroid sülfataz geninin, X kromozomunun inaktif olmayan bir kısmında olmasıyla, liyonizasyon olsa bile fonksiyonuna devam ettiği öne sürülmüştür.
X'e bağlı hastalığı olan bir kadın, mozaik deri lezyonu olsun ya da olmasın, erkek çocuklarına %50 oranında hastalığı tam olarak, kız çocuklarına ise %50 oranında mozaik deri hastalığını veya taşıyıcılığı geçirebilir. X'e bağlı hastalığı olan bir erkek tüm kız çocuklarına mozaik hastalığı veya taşıyıcılığı aktarırken, hiçbir erkek çocuğuna hastalığı aktarmaz.
X kromozomu ile geçen hastalıkların mozaik deri bulguları, hastalığın baskın (dominant) veya çekinik (resesif) kalıtımına göre farklı şekillerde karşımıza çıkabilir.
I. X Kromozomuna Bağlı ve Dominant Geçiş
Bu hastalıklar ne yazık ki erkek çocuklar için yaşamla bağdaşmadığı için sadece kadınlarda görülmekte ve kadın hastalarda fonksiyonel X kromozomu mozaisizmine bağlı olarak çizgisel deri lezyonları şeklinde ortaya çıkmaktadır. Örnek olarak: İnkontinensia pigmenti, fokal dermal hipoplazi (Goltz sendromu), Conradi-Hünermann-Happle sendromu verilebilir.
Nadiren erkek hastalarda Blaschko çizgilerine uyan lezyonlar görülmektedir. Bu durumda erkekte X kromozomu mozaisizmine izin veren bir durum söz konusudur; postzigotik somatik mutasyon, mayoz sırasında yarı-kromatid mutasyon ya da XXY genotipinde ekstra X kromozomunun fonksiyonel (liyonizasyon) mozaismi gibi. X kromozomu mozaisizmi bulunan erkeklerde, eğer mozaisizm gonadları etkilemişse, hastalığı kız çocuklarına geçirebilir.
I. AI. İnkontinensia Pigmenti (Bloch-Sulzberger Sendromu)
İnkontinensia pigmenti (İP), gövde, kol ve bacaklarda çizgisel veya helezonik görünümlü, hiperpigmente, deriden kabarık olmayan (maküllerle karakterize) ve Blaschko çizgilerini takip eden lezyonların görüldüğü bir genodermatozdur. Karakteristik deri bulgularının yanı sıra çeşitli sistemik bulgularla da ilişkili olabilen bir hastalıktır.

I. AII. Goltz Sendromu (Fokal Dermal Hipoplazi)
Goltz Sendromu, nadiren rastlanan bir hastalık olup embriyonal gelişim ve farklılaşma sürecinde ektoderm, mezoderm ve endoderm olmak üzere her üç tabakayı da etkilemektedir. X'e bağlı dominant geçişli olup, erkeklerde doğum öncesi ölümle sonuçlanır. Hastalığın, mozaisizmde liyonizasyonla X inaktivasyonundan kaçan ve mutant aleli taşıyan hücrelerin yıkımına bağlı olarak oluştuğu düşünülmektedir. Bu nedenle, kadınlarda liyonizasyona bağlı olarak Blaschko çizgilerinde lezyonlar şeklinde görülür.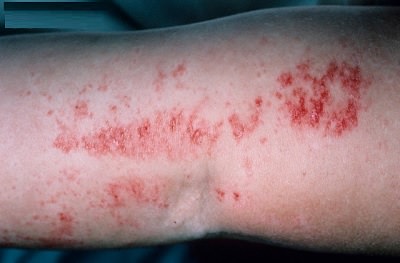
I. AIII. Conradi-Hünermann-Happle Sendromu (CHHS) - X'e Bağlı Dominant Kondrodisplazi Punktata
Conradi-Hünermann-Happle Sendromu, X kromozomuna bağlı dominant geçişli olup, deri, iskelet sistemi ve göz bulgularıyla karakterizedir.
Dermatolojik Bulgular Doğumda, Blaschko çizgilerini izleyen, skuamlı iktiyoziform eritroderma bulguları vardır. Eritroderma genellikle yaşamın ilk yılında kendiliğinden geriler. Ardından, çizgiler ve girdap şeklinde lezyonlarla atrofoderma ve nadiren hiper veya hipopigmentasyon belirir. Saçlı deri tutulumunda ise yama tarzı skatrisyel alopesi görülür.
İskelet Anomalileri
-
Kısa boy
-
Kafa-yüz deformiteleri (frontal çıkıntı, malar hipoplazi ve yassı burun kökü)
-
Asimetrik kol ve bacak eksiklikleri
-
Vertebral malformasyonlar
-
Kalça displazisi
-
Kondrodisplazi punktata bulgusu (epifizlerde noktasal kalsifikasyon)
Göz Bulguları Katarakt, mikroftalmi, mikrokornea, glokom ile birlikte retina ve optik sinir atrofisi görülebilir.
Patojenez CHHS, X kromozomundaki emopamil bağlayıcı protein genindeki mutasyonlar sonucu oluşur. Söz konusu protein, kolesterol biyosentezinde görev yapmakta olup, eksikliğinde kolesterol sentez yolunun ara ürünleri plazma, deri ve diğer dokularda birikir.
I. AIV. CHILD Sendromu (Konjenital Hemidisplazi + İktiyoziform Nevüs + Kol ve Bacaklarda Defektler)
CHILD sendromu, X'e bağlı dominant bir hastalık olup erkeklerde ölümcül sonuçlanmaktadır. Vücudun tam sol ya da sağ yarısında inflamatuar epidermal bir nevüs ile aynı taraftaki iskelet ya da iç organ hipoplazileri veya aplazileri eşlik etmektedir.
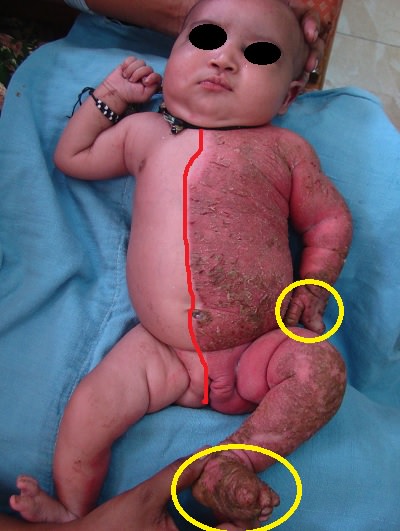
I. AV. MIDAS Sendromu (Küçük Göz - Mikroftalmi ve Çizgisel Deri Defektleri)
MIDAS sendromu, küçük göz yapısı, dermal aplazi ve sklero-kornea bulgularıyla karakterizedir ve genetik ile klinik olarak fokal dermal aplaziden farklıdır. Daha çok yüz ve boyun bölgesinde Blaschko çizgilerini izleyen eritemli dermal aplazik lezyonlar bulunur. X'e bağlı bir hastalık olan MIDAS sendromunun erkeklerde ölümcül olduğu düşünülmektedir, bu nedenle daha çok kız çocuklarında görülür. Hastalık, Xp22 bölgesinde özellikle HCCS genini içeren mutasyonlara bağlı olarak oluşmaktadır. Aile içinde ve aileler arasında fenotipik bulguların çeşitlilik göstermesi, X inaktivasyonundan etkilenen dokuların her bireyde farklı olmasına bağlanmıştır.
I. AVI. Oral - Fasiyal - Dijital Sendrom Tip 1
X'e bağlı dominant geçişli bir hastalık olup; çene ve dilde yarıklar, parmaklarda malformasyon, zeka gelişim geriliği ve Blaschko çizgilerini izleyen dağılımda alopesi alanları ile karakterizedir.
I. B. X Kromozomuna Bağlı ve Resesif Geçiş
Mutasyona uğramış gen erkeklerde ölümcül olmadığı için hastalık erkeklerde görülebilir ve kadınlara göre daha yaygındır. Kadınlar mozaisizmden dolayı daha az etkilenir ve tipik olarak mozaik deri lezyonları görülür. Nadiren X inaktivasyonunun asimetrisi nedeniyle kadınlarda hiç hastalık görülmeyebilir ya da şiddetli hastalık görülebilir. Erkeklerde Klinefelter sendromu olması durumunda, ekstra X kromozomunun liyonizasyonundan kaynaklanan mozaisizm nedeniyle Blaschko çizgileri görülebilir.
I. BI. X'e Bağlı Hipohidrotik Ektodermal Displazi
X'e bağlı resesif kalıtılan hipohidrotik ektodermal displazi, EDA gen mutasyonuna bağlı olarak gelişmektedir. Erkeklerde yaygın hastalık olarak görülürken, kadın taşıyıcılar Blaschko çizgilerini izleyen dağılımda hafif derecede dental anomaliler, hipohidroz, hipotrikoz ve yüzeyde çökük hiperpigmentasyon bulguları gösterebilmektedir.
I. BII. Menkes Sendromu
Menkes Sendromu, X'e bağlı resesif geçişli bir hastalık olup, bakır taşıyıcı ATPaz disfonksiyonuna bağlı oluşur. Kadın taşıyıcılarda, anormal bakır metabolizması gösteren hücrelerin mozaik dağılımına bağlı olduğu düşünülen yama şeklinde saç anomalileri (pili torti) ve Blaschko çizgilerini izleyen hipopigmente lezyonlar görülmektedir.
I. BIII. Partington Sendromu
X kromozomuna bağlı resesif bir hastalık olan Partington sendromu, “Ailesel kutanöz amiloidoz” veya “Partington kutanöz amiloidozu” olarak da bilinir. Kadın taşıyıcılarda çizgisel ve sarmal şeklinde hiperpigmentasyon bulgularına rastlanabilmektedir. Hastalığı bulunan erkeklerde yaygın retiküler hiperpigmentasyon, yüksek yerleşimli frontal saç çizgisi, kronik pulmoner bozukluk, fotofobi ve hipohidroz bulunur.
I. BIV. İmmün Yetmezliğin Eşlik Ettiği Hipohidrotik Ektodermal Displazi
İmmün yetmezliğin eşlik ettiği hipohidrotik ektodermal displazi (İY-HED), inkontinentia pigmenti ile alelik bir genetik hastalık olup NEMO genindeki hipomorfik mutasyonlara bağlı gelişmektedir. Erkeklerde ektodermal displazi ve immün yetmezliğe bağlı tekrarlayan enfeksiyonlarla karakterizedir. Kadın taşıyıcılar ise X inaktivasyonundan etkilenen dokuların dağılımına göre farklı fenotipler gösterebilmektedir. Blaschko çizgileri üzerinde dağılmış hiperpigmente lezyonlara bazı hastalarda immün yetmezlik ve değişken derecede ektodermal displazi bulguları eşlik edebilmektedir.
I. B.V. X'e Bağlı Diskeratozis Konjenita
X'e bağlı diskeratozis konjenita, DKC1 (diskerin proteini) gen mutasyonuna bağlı ortaya çıkmaktadır. Retiküler deri pigmentasyonunun eşlik ettiği atrofi, tırnak distrofisi, mukozal lökoplaki, ilerleyici kemik iliği yetmezliği ve kanserlere artmış eğilim ile karakterizedir. Kadın taşıyıcılarda, mutant genin aktif X kromozomunda olup olmamasına bağlı olarak (X-inaktivasyonundan kaçma), farklı fenotipik özellikler görülebilir. Buna göre, kadın taşıyıcılarda nadiren Blaschko çizgilerini izleyen hiperpigmentasyon veya poikiloderma ile palmoplantar hiperkeratoz bulunabilir.
I. BVI. Alopesi ve Fotofobinin Eşlik Ettiği İktiyozis Follikülaris
Alopesi ve fotofobinin eşlik ettiği iktiyozis follikülaris (IFAP) sendromu, MBTPS2 gen mutasyonuna bağlı olarak ortaya çıkmaktadır. Kadın taşıyıcılarda Blaschko çizgilerini izleyen alopesi, foliküler iktiyoz, atrofoderma ve hipohidroz bulguları görülebilmektedir.