
- Gösterim: 7298
Hipertrikozis, vücudun herhangi bir yerinde kılların sayıca artması ve kalınlaşması anlamına gelir. Sıklıkla, androjen hormonlarına duyarlı bölgelerde aşırı kıl büyümesiyle karakterize olan hirsutizmden ayırt edilmelidir.
-
Çocukluk Çağı: Çocukluk çağında açıklanamayan hirsutizm, genellikle çocuklarda hipertrikoz için endokrin sistemin detaylı araştırılmasını gerektirir.
-
Yayılım: Hipertrikoz, vücudun belli bir bölgesinde lokalize veya tüm vücutta genel olarak ortaya çıkabilir.
-
Başlangıç Yaşı: Kılların aşırı büyümesi, başlangıç yaşına göre doğuştan (konjenital) olabileceği gibi, ileri yaşlarda sonradan (edinsel) da ortaya çıkabilir.
-
İlişkili Durumlar: Hipertrikoz, izole bir bulgu olabilir veya diğer anormalliklerle ilişkili olarak bir sendromun parçası olabilir. Doğuştan hipertrikozlar, neden olduğu estetik problemler nedeniyle çocuğun yanı sıra ebeveynleri de psikolojik olarak etkiler. Ek olarak, hipertrikoz bir sendromun bileşeni olabileceği için erken tanısı ve tüm vücut sistemlerinin multidisipliner değerlendirmesi gereklidir.
Mevcut tedavi seçenekleri sınırlıdır; tatmin edici sonuçlar zaman almakta ve sabır gerektirmektedir.
Lokalize Hipertrikozis
Lokalize hipertrikozis, vücudun belli bir anatomik bölgesinde yaş, ırk veya cinsiyetten bağımsız olarak aşırı kıllanma ile karakterize nadir bir durumdur. Bu durum doğuştan (konjenital) olabileceği gibi sonradan (edinsel) da ortaya çıkabilmektedir.
Lokalize, Sonradan Gelişen (Edinsel) Hipertrikozis
Cildin kronik sürtünme, tahriş ve inflamasyona maruz kalan alanlarında lokal hipertrikozis gelişebilmektedir.
Lokal hipertrikozisin gelişebileceği bildirilen durumlar şunlardır:
-
Dövmeler sonrası.
-
Skleroterapilerde.
-
Kronik venöz yetmezlikte bacaklarda.
-
Derin cilt yanıklarında yanık kenarlarında.
-
Çok kaşıntılı böcek ısırıklarında.
-
Kasıkta lenf bezlerinin alınması sonrası bacakta düzensiz bir desende.
-
Zona yerleşim alanında.
-
Su çiçeği sonrası.
-
Kontakt temas egzamalarında.
-
Ortopedik alçı ve atellerin uzun süre uygulanması sonrası bu alanlarda.
Lokalize, Doğumsal (Konjenital) Hipertrikozis
Lokalize konjenital hipertrikozisler: .
Becker Nevüsü
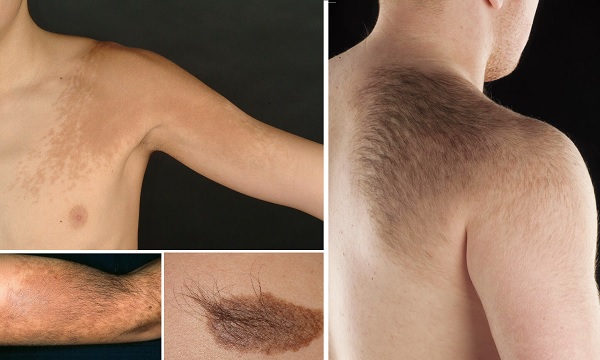
Becker nevüsü, Becker melanozu ve pigmentli kıllı epidermal nevüs olarak da bilinir. Bu durum; doğuştan olup, vücutta belli bir anatomik alana yerleşim gösteren (lokalize), tek taraflı hiperpigmentasyon ve tipik olarak üzerinde aşırı kılların büyümesi (hipertrikozis) ile karakterize bir cilt lekesi olarak görülür.
-
Yaygınlık: Genel olarak 'lik bir yaygınlığa sahiptir ve öncelikle erkekleri etkiler.
-
Nedeni: Artmış androjen duyarlılığından kaynaklandığı düşünülmektedir.
-
Kabul Edilen Durum: Genellikle estetik kaygı dışında zararsız olarak kabul edilir.
-
Ayırıcı Tanı: Ancak, özellikle malign melanom gibi cilt kanserlerinden ayrımının yapılması çok önemlidir.İlişkili Sendrom ve Tedavi
-
Sendrom İlişkisi: Becker melanozisi, Becker nevüs sendromu olarak bilinen ektodermal anormalliklerle ilişkili diğer bulgularla bağlantılı olabilir. Bu yönde hastanın detaylı değerlendirilmesi önemlidir.
-
Tedavi: Hiperpigmentasyon veya hipertrikozis için lazer tedavileri estetik nedenlerle uygulanmaktadır. Daha detaylı bilgi için...
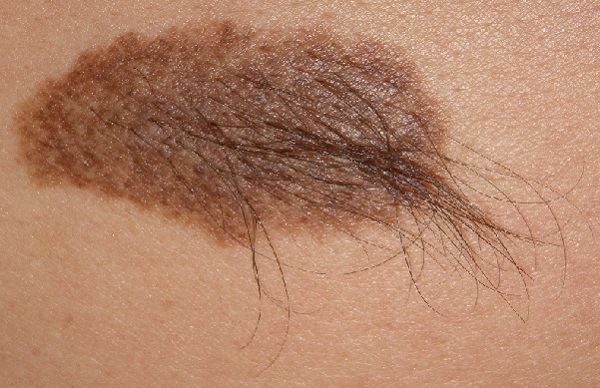
Konjenital Melanositik Nevüsler (Doğumsal Benler)
Konjenital melanositik nevüsler (KMN), doğumsal olan ve üzerinde kıllar taşıyabilen benlerdir. Daha detaylı bilgi için...
Özellikle dev boyutlu formları ($\mathbf{10\ \text{cm}^2$'den büyükler}$), kanserojen riskleri nedeniyle önemlidir:
-
Dev KMN'lerde kanser riski 'tür.
-
Daha küçük KMN'lerde ise kanser riski oranında bildirilmiştir.
Dev boyutlu KMN'lerde doğuştan benler üzerinde hipertrikozis (aşırı kıllanma) mevcuttur ve bu kılların artışı ergenlikte daha belirgin hâle gelmektedir. Kıllar neredeyse saça benzer formdadır.
Bu tür benler, aşağıdaki sistemik ve nörokutanöz problemlerle birlikte olabilir:
-
Göz problemleri
-
Nörofibromatozis
-
Kemik malformasyonları
-
Leptomeningeal problemler (Nörokutanöz melanoblastozis)
-
Hidrosefali
-
Oligofreni
Nevoid Hipertrikozisler
Nevoid hipertrikozisler, nevoid (benlere benzer) ancak pigmentasyon değişimlerinin olmadığı aşırı kıllanma durumlarıdır. Bunlar, vücutta lokal yerleşimli terminal kıl büyümesi ile karakterize nadir görülen konjenital hastalıklardır. Cilt lezyonları genellikle doğumda veya hemen sonrasında mevcuttur. Lezyonlar tipik olarak tektir; ancak ergenlikten sonra gelişen çoklu lezyon vakaları da bildirilmiştir. Çoklu lezyonlar sıklıkla "Blaschko çizgileri" boyunca yuvarlak, dama tahtası veya doğrusal desenler şeklinde görülmektedir. Saçlı deri yerleşimlerinde terminal hipertrikozis gösteren saçlar, orijinal saç renginin aynısı olabilir; nadiren gri veya beyaz olabilmektedir. Nevoid hipertrikozların etiyolojisi tam olarak bilinmemektedir. Genetik geçişler gösterebildiği gibi, bir malformasyonun parçası da olabilmektedir. Hiçbir neden ya da anomaliye bağlı olmaksızın nevoid sınırlı hipertrikozun en basit örneği, tek başına ve çok gür bir kaşın varlığıdır. Nevoid hipertrikozlar, anatomik yerleşim yerlerine ve birlikte olabilecek patolojilere göre aşağıdaki gibi tanımlanmıştır:
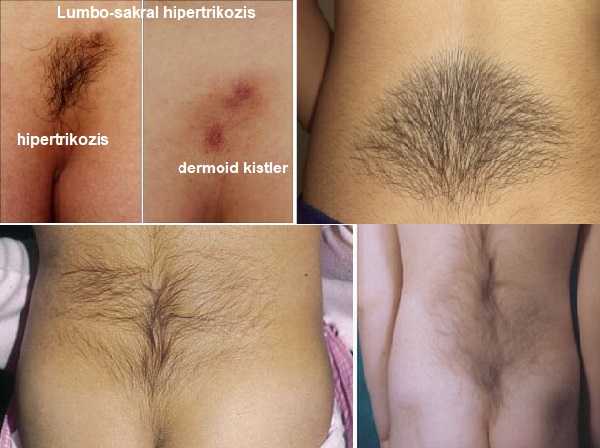
- Lumbosakral Hipertrikozis (Faun Kuyruğu Deformitesi): Lumbosakral hipertrikozis, Faun kuyruğu (Faun tail) deformitesi olarak da bilinmektedir. Sırt orta hattında, altta sakral alanda lokalize kıllanma artışı olarak tanımlanmıştır.Hipertrikozis, çeşitli eş zamanlı cilt malformasyonları ile ilişkilidir. Bunlar: Sakral gamze, hiperpigmentasyon, lipom, port şarabı lekesi ve dermoid kisttir. Bu gelişimsel defekt genellikle kemik ve omurilik defektleriyle birliktedir; en sık görülenler spina bifida okkulta ve diastematomiyeli'dir. Bu nedenle hasta mutlaka radyolojik ve nörolojik problemler açısından değerlendirilmelidir.Tespit edilmez ve cerrahi olarak düzeltilmezse, ilerleyen yaşla birlikte komplikasyonlar ortaya çıkabilir. Vertebraların omurgaya göre orantısız bir şekilde büyümesi nörolojik anormalliklere neden olabilir.En yaygın kusurlar ayak düşüklüğü, gece idrar kaçırma ve sırt ağrısıdır
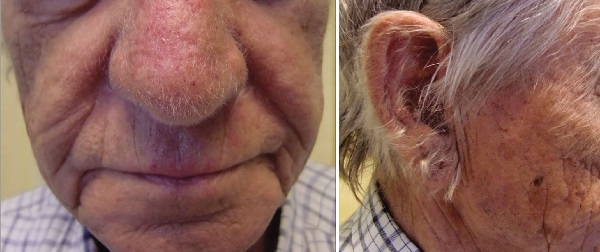
- Kulakta Hipertrikozis (Hipertrikozis Pinnae Auris / Kıllı Kulaklar): İnsan kulağının kulak kepçesinde aşırı sert siyah kılların varlığı, hipertrikozis pinnae auris veya kıllı kulaklar olarak adlandırılır. Kulaklarda kıllanma öncelikle erkeklerde, ara sıra kadınlarda yaşlanma ile birlikte görülmektedir. Hintlilerde ırksal olarak en fazla görülür. 'li yaşlarda başlamakta ve 'li yaşlarda en belirgin olduğu dönemdir. Çalışmalar hipertrikozis pinnae auris'in Y kromozomu ile ilişkili olduğunu ve kalıtımının babadan oğula geçtiğini göstermektedir. kromozomal anomalilerde de kulakta kıllanma olmaktadır. Diyabetik annelerden doğan çocuklarda kulak kılları artışı gözlenebilmektedir. Sonradan gelişen kulak kılı artışlarında AIDS düşünülmelidir.
- Burunda Hipertrikozis (Hipertrikozis Nasi / Kıllı Burun): İnsan burnunda aşırı sert siyah kılların varlığı hipertrikozis nasi veya kıllı burun olarak adlandırılır.
- Kaş Ortası Hipertrikozis: Sıklıkla aileseldir. Ayrıca siklosporin ilaç kullanımında ve AIDS'te kaş ortası kıllanma artışı olabilmektedir.
- Hipertrikozis Kübiti (Kıllı Dirsek): Doğuştan dirsek çevresinde hipertrikozun varlığı ile tanımlanır. Hipertrikoz, dirsekten ön kol ve üst kola dağılım gösterebilmektedir. Bazı vakalarda açıklanamayan kısa boy ile birlikte kolların dış yüzeyinde uzun vellus kıllar ve hipertrikoz görülebilmektedir; bu durum "tüylü dirsek sendromu" olarak tanımlanmaktadır. Bu sendromda ince lanugo kıllar, kolların doğumda simetrik olarak bulunur.
- Kıllı Aksesuar Meme (Hairy Polythelia): Aksesuar meme dokusu ve hipertrikozisi tanımlar. Bazen meme dokusu olmadan sadece hipertrikozis olabilmektedir. Ailesel olanlarda ektopik böbrek (mammo-renal sendrom) olabilmektedir. Son zamanlarda hairy polythelia ile birlikte testis, mesane, prostat ve böbrek karsinoması ile birliktelikleri bildirilmiştir.
- Anterior Servikal Hipertrikozis (Boyun Ön Kısmı): Boyun ön kısmında küçük bir alanda aşırı kıllanma vardır. Hipertrikoz, çene altı ile dekolte bölgesi arasında boyun ön kısmında yer almaktadır. Kalıtım şekli büyük olasılıkla otozomal resesiftir. Genellikle izole bir bulgu olmasına rağmen, periferik duyusal ve motor nöropati ile ilişkili olabilir.
- Posterior Servikal Hipertrikozis (Boyun Arka Kısmı): Aşırı kıllanma boynun arka kısmında, servikal omurların üzerinde bulunur. Ailesel olabilmektedir. X'e bağlı veya otozomal dominant bir özellik olarak tanımlanmıştır. Altta kifoskolyoz veya kamburluk bulunabilmektedir.
- Saçlı Deride Nevoid Hipertrikozis: Saçlı deride ortada küçük alopesi alanı veya alopesi olmadan terminal saçlarda hipertrikozis ile karakterizedir. Saçlar normalden koyu olabilir. Merkezi sinir sistemi anomalileri açısından değerlendirilmelidir.
Tüm Vücutta Yaygın (Genel) Hipertrikozis
Vücutta yaygın hipertrikozis, doğumsal olarak ya da yaşamın herhangi bir evresinde sporadik olarak ortaya çıkabilmektedir.
Akiz (Sonradan Gelişen, Edinilmiş) Genel Hipertrikozis
Vücutta yaygın hipertrikozis, yaşamın herhangi bir evresinde ortaya çıkabilir. Ergenlik öncesi altta hiçbir patolojiye bağlı olmadan görülebileceği gibi, paraneoplastik sendromlar gibi ciddi bir patolojiye ikincil olarak da ortaya çıkabilmektedir.
- Akiz Hipertrikozis Lanuginosa: Bu durum, özellikle "paraneoplastik dermatoz" olarak bilinmektedir. Çoğu zaman altta yatan bir kanser ile ilişkilidir. Akciğer, kalın bağırsak, prostat, yumurtalık, rahim, pankreas, rahim ağzı kanserleri, lenfoma ve lösemi ile birlikteliği vardır. Bu klinik tablonun varlığı, altta yatan kanserin kötü seyredeceğini göstermektedir ve kanser belirtilerinden daha önce görülebilmektedir. Hipertrikoziste ince ve daha açık renkli lanugolar görülür. Lanugolar haftada olacak şekilde hızlı bir şekilde uzayabilir ve 'ye kadar ulaşabilir. Lanugolar yüzü kaplayacak şekilde tüm vücutta görülebilir.
- Universal Hipertrikozis: Bu formun genetik olduğu bilinmektedir. Burada kılların vücutta dağılımı normaldir ancak sayıca çok fazladır. Kılların kalınlıkları ve uzunlukları yaşla hızla artmaktadır.
- Prepubertal Hipertrikozis: Bu durum, ergenlik dönemi öncesi sağlıklı çocuklarda gözlenmektedir. Tüm vücutta, ancak özellikle yüz, kol ve bacakların üst kısmında ve sırtta kıllar fazladır.
- İatrojenik Hipertrikozis (İlaç Kaynaklı): Edinilmiş genel hipertrikozda, özellikle ilaç kullanımına bağlı olanlar, iyatrojenik hipertrikozis olarak tanımlanır. İlaçlar genel hipertrikozislerin en yaygın görülen birincil nedenleri arasında yer almaktadır. Bu nedenle, sonradan gelişen ve genel hipertrikoziste ilaç kullanımı mutlaka sorgulanmalıdır. Birkaç ilacın, özellikle oral minoksidil, diazoksit, fenitoin sodyum ve siklosporinin, yaygın hipertrikoza neden olduğu iyi bilinmektedir. Her ilaç hipertrikozisi benzeriz özellikleri ile göstermektedir. Örneğin, Minoksidil öncelikle yüz, omuzlar, kol ve bacaklarda hipertrikoza neden olur. Ek olarak, oral kontraseptifler, sistemik kortikosteroidler ve bazen psoralenler ile streptomisin sülfat gibi diğer ilaçlar da hipertrikozise neden olmaktadır. İlaç kullanımının kesilmesi, hipertrikozisin kendiliğinden kaybolmasına yol açar.
- Bazı Sistemik Hastalıklara Bağlı Hipertrikozis: Yaygın hipertrikozis, bazı sistemik hastalıklarla da ilişkilidir. Bunlara örnek olarak siringomyeli, diabetes mellitus, HIV/AIDS, hipotiroidizm, pretibial miksödem ve fetal alkol sendromu verilebilir.
Doğumsal (Konjenital) Genel Hipertrikozis
Doğumsal genel hipertrikozis, mukozalar, avuç içleri, ayak tabanları, sünnet derisi ve glans penis gibi sadece kıl olmayan bölgeler hariç, tüm vücut yüzeyinde dikkate değer miktarda aşırı kıllanma bulunmasıyla karakterizedir. Bu klinik grup, bir sendrom ile birlikte olabileceği gibi, sendromlara bağlı olmaksızın izole olarak da ortaya çıkabilmektedir.
İzole Doğumsal Genel Hipertrikozis
- Hipertrikozis Lanuginosa Konjenita: Hastaların 'sinde otozomal dominant geçiş söz konusudur; 'ünde ise genetik geçiş tam olarak bilinmiyor. Doğuştan lanugo kılların olduğu hipertrikozis tüm vücutta (mukozalar, avuç içleri ve ayak tabanları hariç) bulunmasıyla karakterize nadir bir hastalıktır. Sarı ile siyah lanugo kıllar doğumda mevcut olabilir veya bebeklik döneminde gelişebilir. Sonradan gelişen klinik formda ise hipertrikozis yaşlarında başlayabilmektedir. Lanugolar sıklıkla sırtta vertebra üzerinde ve özellikle lumbosakral (bel) bölgesinde yer almaktadır. Ayrıca kulak ve çevresinde de bulunmaktadır. Yaşla beraber gövdede lanugolar azalırken yüzdekiler artmaktadır. Klinik tablo tek başına estetik bir problem olmakla birlikte diş gelişim problemleri (erken çıkması ya da olmaması gibi), kulak defektleri, sindirim sisteminde pilor stenozu, iskelet deformiteleri, gözde katarakt ve fotofobi ile birlikte olabilmektedir.
- Gingival Fibromatozis ile Birlikte Hipertrikozis Lanuginosa Konjenita: Hipertrikozis, bu çocuklarda genellikle hipertrikozis lanuginosa ile aynı dağılım göstererek tüm vücutta ve yüzde bulunmaktadır. Hipertrikozis sıklıkla doğumda mevcuttur veya erken bebeklik döneminde gelişir, ancak hastaların yarısında hipertrikozis ergenlik döneminde başlar. Bazı çalışmalarda bu hasta grubunda lanugo yerine terminal kıllar tanımlanmıştır. Otozomal dominanttır. Diş etlerinde büyüme (gingival hiperplazi) genellikle dişler beklenen programa göre çıkmadığında ve dolayısıyla genellikle hipertrikozis gözlemini takiben fark edilir. Diş etleri pembe, sert, kaba ve nodüler görünümdedir. Dişlerin çıkmaması periodontal apselerle ilişkili olabilir. Gingival hiperplazi ve hipertrikozise ek olarak zihinsel gerilik ve/veya epilepsi nöbetleri görülebilmektedir. Bu hastalardaki komplikasyonlar arasında çiğneme, solunum ve konuşmada bozulma bulunur.
- Doğumsal Genel Hipertrikozis: Kalıtım: kromozomunda X'e bağlı dominant geçiş söz konusudur. Doğuştan görülen hipertrikozis terminal kılları içermektedir. Cinsiyet Farkı: Erkekler kadınlardan daha sık etkilenir. Etkilenen erkeklerde terminal kılların tüm vücutta genel bir dağılımı vardır. Kadınlarda hipertrikozis asimetrik ve yamalı bir yapıdadır.
Sendromlar ile Birlikte Görülen Doğumsal Genel Hipertrikozis
Bu doğumsal ve tüm vücutta genel görülen hipertrikozis, bazı sendromlar ile birlikte olduğu için önemlidir. Sendromlar ile birlikte olabilen bu hipertrikozlar, bir kromozomal anomaliye bağlı olarak ortaya çıkabildiği gibi genetik geçişli de olabilmektedir.
- Kromozomal Anomaliye Bağlı Sendromlar: Kısmi Trizomi bu grupta yer almaktadır ve nadiren bildirilen bir kromozomal bozukluktur. Üçüncü kromozomun uzun kolunun bir kısmının çoğalması ( duplikasyonu). Hipertrikozise ve tipik yüz-kafa şekil bozukluğuna (belirgin alın, epikantik kıvrım, geniş ve düz burun kökü, öne dönmüş ve belirgin burun delikleri, belirgin filtrum, mikrognati, yarık dudak ve yarık damak, ağız köşelerinin aşağı kıvrık olması ve ince dudaklar, kulaklarda şekil bozukluğu ve kısa boyun), Kistik higromaya, beyin malformasyonlarına (mikrosefali ve Dandy-Walker malformasyonu), Göz malformasyonlarına (şaşılık, nistagmus, kornea opasiteleri, katarakt, iris kolobomları, optik sinir kolobomu ve anoftalmi), Konjenital kalp defektlerine (kalp septal defektleri), Böbrek malformasyonlarına (polikistik böbrekler veya displazi) ve iskelet anomalilerine (falanksların hipoplazisi, kamptodaktili ve klinodaktili) yol açar.
- Kromozomal, genetik geçişli sendromlar ile birlikte görülen doğumsal genel hipertrikozis; Genetik geçiş, otozomal dominant, otozomal resesif ya da X'e bağlı geçişe göre alt gruplara ayrılmaktadır.
- Otozomal dominant geçişli sendromlar ile birlikte görülen doğumsal genel hipertrikozis;
- Barber-Say sendromu; özellikle sırtta şiddetli hipertrikozis, seyrek kaşlar ve kirpikler, hiperlaks ve atrofik cilt ile düşük frontal saç çizgisi, oküler telekantus, hipoplastik göz kapakları, periorbital dolgunluk, ektropion, geniş burun köprüsü, soğanlı burun, hipoplastik burun kanatları, kalın geniş burun kanatları, öne dönmüş ve belirgin burun delikleri, belirgin yanak pedleri, büyük ağız, ince üst vermilion sınırı, küçük çene, düşük ve daha arkaya rotasyon gösteren kulaklar, küçük /olmayan meme bezleri ile karakterize nadir bir konjenital hastalıktır.
- Cantu sendromu; hipertrikotik osteokondrodisplazi olarak da bilinen Cantu sendromu, generalize hipertrikoz, lenfödem, fetal parmak pedleri, yumuşak "hamur benzeri" cilt, derin tırnaklar, makrosefali, kaba yüz, hipertelorizm, uzun palpebral fissürler, epikantal kıvrımlar, düz burun köprüsü, uzun filtrum, geniş ağız, kalın dudaklar ve hafif zihinsel engellilik ile karakterizedir. Kardiyak bulgular arasında patent duktus arteriosus, septal hipertrofi, pulmoner hipertansiyon ve perikardiyal efüzyon bulunur. İskelet anormallikleri arasında kalınlaşmış kalvaryum, dar toraks, geniş kaburgalar, düzleştirilmiş veya oval vertebral gövdeler, koksa valga, osteopeni, genişlemiş medüller kanallar ve uzun kemiklerin metafizyel genişlemesi yer alır.
- Tabut-Siris sendromu; Coffin-Siris sendromunun tipik özellikleri arasında kafa derisi hipotrikozu ile vücutta genel hipertrikoz, beşinci parmak ve ayak tırnaklarının yokluğu, beşinci parmakların ve ikinci ila beşinci ayak parmaklarının distal falankslarının yokluğu, kasık fıtığı, emme ve beslenme zorluğu, tekrarlayan üst ve alt solunum yolu enfeksiyonları, azalmış fetal aktivite ve intrauterin büyüme geriliği bulunur. Bu sendromla ilişkili diğer özellikler arasında mikrosefali, gür kaşlar, uzun kirpikler, alçak, düz burun köprüsü, geniş yukarı kalkık burun ucu, kısa filtrum, geniş ağız, belirgin dudaklar ve yarık damak yer alır. Doğum sonrası büyüme geriliği ve orta düzeyde gelişimsel gecikme, daha büyük çocuklarda ve yetişkinlerde yaygın özelliklerdir.
- Kraniofasiyal disostoz-hipertrikoz/Fontaine progeroid sendromu/Gorlin-Chaudhry-Moss sendromu; belirgin yaygın hipertrikoz, doğum öncesi ve sonrası büyüme geriliği, kranial malformasyon (kraniosinostoz), yüz malformasyonu (alçak saçlı deri çizgisi, geniş ve açık ön fontanel, üçgen yüz, orta yüz hipoplazisi, küçük gözler, aşağı eğik palpebral fissürler, dışbükey ve geniş burun köprüsü), yüksek kemerli ve dar damak, dental maloklüzyon, anormal şekilli dişler, oligodonti, mikrodonti, mikrognati, genital hipoplazi ve kalıcı açık duktus ile karakterizedir.
- Cornelia de Lange sendromu; synophrys (medial kaşların orta hatta birleşmesi) ve/veya kalın kaşlar, kısa burun, çukur burun sırtı ve yukarı doğru kıvrık burun ucu, uzun ve/veya düz filtrum, üst dudakta ince kırmızılık ve/veya ağız köşelerinin aşağı doğru kıvrılması, el oligodaktilisi (bir veya daha fazla parmağın doğuştan yokluğu) ve/veya adaktili (tüm parmakların ve/veya ayak parmaklarının yokluğu), doğuştan diyafram fıtığı, gelişimsel gecikme ve/veya zihinsel engellilik, doğum öncesi büyüme geriliği, doğum sonrası büyüme geriliği, mikrosefali (doğum öncesi ve/veya doğum sonrası), küçük eller ve/veya ayaklar, kısa beşinci parmak ve hirsutizm ile karakterizedir.
- Hipertrikoz universalis konjenita/Ambras tipi; yüz, kulaklar ve omuzlarda hipertrikozis, alın, göz kapakları, yanak, burun ve preauriküler bölge, birkaç santimetre uzunluğa ulaşan düzgün bir şekilde kıllarla kaplıdır. Diğer özellikler arasında yüz dismorfizmi (üçgen kaba yüz, gür kaşlar, uzun kirpikler, telekantus, uzun palpebral fissür, çökük burun köprüsü, soğan biçimli burun, yuvarlak burun ucu, antevert burun delikleri, kalın burun kanatları, uzun belirgin filtrum, ince kırmızı kenar ve trapez ağız), iskelet anormallikleri (eksiyal post-aksiyel rudimenter heksadaktili, ekzostoz ve birinci metakarpal kemiğin düşük yerleşimi) ve aksesuar meme uçları ile diş anormallikleri (oligodonti, gecikmiş birincil/ikincil dişlenme) bulunur.
- Lenfödem-distikiyazis sendromu; geç çocukluk veya ergenlikte ortaya çıkan ve genellikle asimetrik olan alt ekstremite lenfödemi ile karakterizedir. Etkilenen bireylerin %94'ünde doğumda mevcut olabilen distikiyazis (meibomian bezlerinden kaynaklanan anormal kirpikler) görülür. Bireylerin yaklaşık %75'inde kornea hasarı, tekrarlayan konjonktivit ve fotofobi vardır. Diğer yaygın bulgular arasında varisli damarlar, konjenital kalp hastalığı ve ptozis bulunur.
- Rubinstein-Taybi sendromu; zihinsel engellilik, doğum sonrası büyüme geriliği, yüzün çarpıcı dismorfik özellikleri (mikrosefali, yüksek kemerli kaşlar, uzun kirpikler, aşağı eğik palpebral fissür, geniş burun köprüsü, gagalı burun, sarkık kolumella, yüksek kemerli damak, yüz buruşturma ve mikrognati), eller ve ayaklar (geniş başparmak ve halluks) ile karakterizedir. Nazolakrimal kanal tıkanıklığı, ptozis, konjenital veya juvenil glokom ve refraktif hatalar gibi oküler anomaliler ve dişlerde sıkışıklık, alt ve üst çenede çapraz kapanış ve tornavida gibi kesici dişler gibi orodental özellikler nadirdir. Rubinstein-Taybi sendromu hastalarında keloidler ve nöral veya gelişimsel kökenli tümörler gelişmeye eğilimlidir.
- Schinzel-Giedion sendromu; bu sendromunun tanısı klinik bulgulara ve/veya SETBP1 mutasyonuna dayanır. Tip 1 (kompleks ve klasik tip) gelişimsel gecikme ve hidronefroz veya karakteristik iskelet anomalilerinden ikisi (sklerotik kafa tabanı, geniş kaburgalar, artmış kortikal yoğunluk ve geniş oksipital senkondroz) ile ilişkili tipik yüz özellikleri (belirgin alın, orta yüz geri çekilmesi, kısa ve yukarı kalkık burun) ile ortaya çıkar. Tip 2 (orta tip) gelişimsel gecikme, belirgin yüz fenotipi gösterir ancak hidronefroz ve SETBP1 mutasyonunun varlığı ile iskelet anomalileri yoktur. SETBP1 mutasyonu olan tip 3 (basit tip) ifade edici dil gecikmesinin çarpıcı özellik olduğu gelişimsel gecikme ile ortaya çıkar. Hipertrikoz genellikle bebeklikte kaybolan bir özellik olabilir.
- Sert cilt sendromu; kalça ve uylukların üzerindeki cildin kaya gibi sertleşmesi, üstteki cildin hipertrikozisi ve hiperpigmentasyonu ile ortaya çıkar. Ekstrakutanöz özellikler arasında büyük eklem kontraktürleri, skolyoz, ayak ucunda yürüyüş, dar toraks, büyüme geriliği ve kısıtlayıcı akciğer hastalığı bulunur.
- Weidemann-Steiner sendromu; dismorfik yüz hatları (kalın kaşlar, uzun kirpikler, aşağı eğik palpebral fissürler, geniş burun köprüsü, geniş burun ucu, ince üst dudak ve mikrognati), hipertrikoz, kısa boy, gelişimsel gecikme ve zihinsel engellilikle ilişkilidir. Serebral, kardiyak, renal ve optik yapıların konjenital malformasyonları da bildirilmiştir.
- Zimmermann-Laband sendromu; başlıca klinik özellikleri diş eti büyümesi, soğan biçimli yumuşak burun, kalın ve sarkık kulaklar, tırnak aplazisi veya hipoplazisi, hipertrikoz, eklem hiperekstansiyonu, hepatosplenomegali ve epilepsi ile veya epilepsi olmaksızın zihinsel engelliliktir.
- Otozomal resesfi geçişli sendromlar ile birlikte görülen doğumsal genel hipertrikozis;
- Leprechaunizm/Donohue sendromu ve Rabson-Mendenhall sendromu; İnsülin reseptörü geni INSR'deki mutasyonlar, kalıtsal insülin direnci sendromlarının bir yelpazesine neden olur. Bu bebeklerde bir yıllık yaşam ve sonrasında ölümle en şiddetli fenotipi gösteren leprechaunizm sednromu yer alırken bir yıldan uzun yaşamla ancak genellikle ergenlikten önce ölümle sonuçlanan ara bir fenotip olan Rabson-Mendenhall sendromu ve daha hafif formu genellikle ergenlikten sonra belirginleşen A tipi insülin direnci. Hem leprechaunizm hem de Rabson-Mendenhall sendromu, belirgin kraniyofasiyal özellikler (küçük elf yüzü, büyük düşük kulaklar, belirgin gözler, geniş burun delikleri, kalın dudaklar ve büyük ağız), hipertrikozis, akantozis nigrikans, lipoatrofi, doğum öncesi ve sonrası büyüme geriliği, erken ergenlik, meme hiperplazisi, belirgin meme uçları, şişkin karın, büyük eller ve ayaklar, kas atrofisi, penis büyümesi, klitoromegali ve polikistik overler ile karakterize benzer fenotipik belirtilere sahiptir. Ancak Rabson-Mendenhall sendromu, leprechaunizmden daha az şiddetli olması, erken ve displazik dişlenme, kaba yüz görünümü, diş eti hiperplazisi, epifiz hiperplazisi ve bir yaşından sonra hayatta kalma gibi belirtilerle farklılık gösterir.
- Doğuştan yaygın lipodistrofi/Berardinelli-Seip sendromu; hipertrigliseridemi, kas hipertrofisi, hepatik steatoz (karaciğer yetmezliği nedeniyle siroza ve ölüme ilerleyebilir), diabetes mellitus, akut pankreatit, erken kardiyovasküler hastalık, akantozis nigrikans ve iştah artışı (leptin ve adiponektin gibi adipositokinlerin düşük serum seviyeleri nedeniyle) gibi insülin direncinin belirti ve semptomlarıyla karakterizedir. Çocukluk döneminde insülin benzeri büyüme faktörü 1 hipersekresyonu nedeniyle mandibula, el ve ayaklarda hafif büyüme ile birlikte bir akromegaloid görünüm görülür. Ergenlikte, insülin direnci kadınlarda polikistik over sendromu riskini artırırken erkeklerde normal doğurganlık vardır.
- Doğuştan eritropoietik porfiri (Gunther hastalığı); hastalık, erken fotosensitivitenin eritem, kabarcıklar ve erozyonlara yol açmasıyla karakterizedir ve skarlar, yara izi bırakır. Kemik rezorpsiyonu ile sekonder enfeksiyon, parmakların, ayak parmaklarının ve burun, kulaklar ve göz kapakları dahil olmak üzere yüz özelliklerinin deformitesine ve şekil bozukluğuna yol açabilir. Ekstremitelerde fokal hipo veya hiperpigmentasyon ve hipertrikozis meydana gelebilir. Dişlerin minesinde ve dentinde porfirin birikimi, güneş ışığında kirli sarı veya kahverengi bir renk ve Wood lambası altında parlak kırmızı bir renk ile kırmızı idrar, kemik kaybı, ektropion, keratokonjunktivit, katarakt ve körlük veren "eritrodonti" ile sonuçlanır. Tüm hastalar hemolitik anemi ve splenomegali ile başvurur.
- Dişeti hiperplazisi olan veya olmayan yaygın hipertrikozis üniversalis; bu durum doğumda veya erken bebeklik döneminde konjenital genel hipertrikozis ile karakterizedir, ancak vakaların yarısında ergenlik döneminde başlar ve çoğunlukla yüzde, üst ekstremitelerde ve sırtın ortasında kendini gösterir. Dişeti hiperplazisi neredeyse her zaman 10 yaşından sonra görülür. Aşağı eğimli palpebral fissürler, epikantal kıvrım, burnun soğanlı ucu, kalın alae nasi, geniş burun kökü ve uzun belirgin veya derin filtrum gibi dismorfik özellikler nadir hastalarda belgelenmiştir.
- Histiyositoz lenfadenopati artı sendromu (H sendromu); H sendromunun temel cilt özellikleri arasında, esas olarak bacaklarda ve alt karında lokalize olan hiperpigmentasyon ve hipertrikozis ile ilerleyici simetrik deride sertleşme bulunur. Temel cilt dışı özellikler arasında hepatosplenomegali, sensörinöral işitme kaybı, hiperglisemi (otoimmün olmayan diabetes mellitus), kalp anomalileri, hipogonadotropik hipogonadizm, kısa boy ve halluks valgus/fleksiyon kontraktürleri olarak ortaya çıkan büyüme hormonu eksikliği bulunur. Erkek deneklerde jinekomasti, skrotal kitleler ve azospermi olduğu bildirilmiştir.
- Lleigh sendromu; merkezi sinir sisteminde ilerleyici psikomotor gerilik, nistagmus, oftalmoparezi, optik atrofi, ataksi, distoni ve solunum yetmezliği olarak ortaya çıkar. Bazı hastalarda polinöropati veya miyopati, nörolojik olmayan anormallikler ve hipertrikozis gibi periferik sinir sistemi tutulumu görülür.
- Oliver-McFarlane sendromu; korioretinopati hipofiz disfonksiyon sendromu olarak da adlandırılan bu durum, tedavi edilmemiş tiroid ve büyüme hormonu anormallikleri, retinitis pigmentosa ve trikomegali sonucu oluşan fiziksel ve zihinsel engellilikle karakterizedir. Hastaların yarısında doğuştan hipogonadizm görülür ve hemen hemen hepsinde ergenlik döneminde belgelenmiş hipogonadotropik hipogonadizm ve sonrasında üreme disfonksiyonu vardır.
- Mukopolisakkaridozlar( X'e bağlı resesif bir desene sahip olan mukopolisakkaridozlar II veya Hunter sendromu hariç); altta yatan mutasyona ve buna bağlı enzim aktivitesinin derecesine, başlangıç yaşına (daha erken başlangıçlılarda daha şiddetli hastalık), sistemik tutuluma ve klinik gidişata göre şu anda yedi tip bilinmektedir.
- Winchester sendromu; iskeletsel radyolojik özellikler (multifokal osteoporoz, ilerleyici osteoliz ve vertebra gövdelerinde, karpal ve tarsal kemiklerde, metafiz ve epifizlerde dejeneratif değişiklikler) ve aşağıdaki özelliklerden en az ikisinin bulunmasıyla karakterizedir: kısa boy ve ilerleyici eklem kontraktürleri, kornea opasiteleri, kösele gibi deri veya hipertrikoz, diş etlerinin hipertrofisi ve kaba yüz hatları.
- Otozomal X geni geçişli sendromlar ile birlikte görülen doğumsal genel hipertrikozis; bu gurupta sadece Cornelia Lange sendromu ve mukopolisakkaridozlar tip II yer almaktadır.
- Otozomal dominant geçişli sendromlar ile birlikte görülen doğumsal genel hipertrikozis;
Hipertrikozisin Gelişiminde Olası Aday Genler ve Genetik Değerlendirme
Hipertrikozda altta yatan mekanizma belirsiz olsa da, saç büyümesini etkileyen birkaç aday gen bulunmaktadır.
-
Angora Geni ():
-
Angora () geninin, saç/kılların büyümesinde düzenleyici bir işlevi olduğu gösterilmiştir.
-
çekinik mutasyonu, anagen ve katagen saç arasındaki geçişi geciktirerek farelerde anormal derecede uzun saç üretir.
-
Bu genin, kıl foliküllerinde bir sinyal molekülü olan fibroblast büyüme faktörü () 'in mutant bir aleli olduğu gösterilmiştir. Henüz hiçbir insanda homologu bulunamamıştır.
-
-
Keratinosit Büyüme Faktörü ():
-
olarak da bilinen keratinosit büyüme faktörünün, saç folikülleri ve yağ bezleri içindeki progenitör hücrelerin çoğalmasını ve farklılaşmasını uyardığı gösterilmiştir.
-
Bu faktörün olmadığı farelerde yağlı veya keçeleşmiş sert kıllar ortaya çıkmaktadır. İnsanlarda keratinosit büyüme faktörünün aşırı salınımı veya mutasyonlarının fenotipik sonucu bilinmemektedir.
-
Bu belirsizliğe rağmen, hipertrikozisli hastalarda mutlaka genetik değerlendirme yapılmalıdır.
-
Tanı Yöntemleri: Moleküler genetik testler, ilişkili genetik etiyolojinin teşhisi ve karakterizasyonu için tercih edilen yöntemdir. Bazı durumlarda, örneğin mukopolisakkaridozlar için lizozomal enzim testleri gibi diğer doğrulayıcı testler istenebilir.
-
Genetik Danışmanlık: Hipertrikozlu bireyin doğru teşhisinin ardından, hastaya ve aileye hastalığın doğası, doğal seyri ve mevcut tedavi seçenekleri ile hastalığın takip planı hakkında uygun genetik danışmanlık sağlanmalıdır.
-
Multidisipliner Yönetim: Hipertrikozla ilişkili genetik sendromların çoğu, yukarıda belirtildiği gibi çoklu sistemik/organ tutulumuna sahiptir ve bu nedenle multidisipliner bir yönetim gerektirir.
-
Ailesel Risk: Tanımlanan genetik etiyolojiye dayanarak, kalıtım örüntüsü (otozomal dominant, otozomal resesif veya bağlantılı olup olmadığı) belirlenebilir ve ailede sonraki gebeliklerde tekrarlama riski hesaplanabilir.
-
Doğum Öncesi Tanı: Etkilenen taşıyıcı ebeveynlerde patojenik genetik varyantlar belirlendikten sonra, mutasyon analizi yoluyla riskli gebelikler için doğum öncesi tanı sunulabilir.
Hipertrikozis Tedavisi Yaklaşımları ve Öneriler
Hipertrikozda kılların bir ömür boyu periyodik olarak alınması zordur. Tedavi gerekliliği; hastanın yaşına, kıllanmanın anatomik yerleşim yerine ve derecesine, ayrıca çocuğun psikososyal ihtiyaçlarına bağlıdır. Aşırı kıllanma yaşayan yenidoğan ve bebeklerde bile, ailenin ve toplumun kabulünü kolaylaştırmak için bazı bölgelerdeki kılların alınması gerekebilir; zira ebeveynler utanç ve sıkıntı duyguları yaşayabilmektedir. Günümüzde hipertrikoz için güvenilir, radikal ve standartları belirlenmiş bir tedavi protokolü maalesef yoktur.
Mevcut tedavi yöntemleri arasında kılların kısaltılması, ağartılması, ağda, kimyasal tüy dökücüler, elektroliz ve lazer epilasyon yer almaktadır.
Geçici Kıl Alma Yöntemleri
Kılların boylarının kısaltılması, kılları daha az belirgin yaptığı için özellikle küçük çocuklar için en çok önerilen seçenektir. Ancak kıllar kesildikçe uçları daha kalınlaştığı için, uygulamalar günlük olarak yapılmalıdır. Makas ile kesme ve trimming sistemleri en güvenli yöntemler arasındadır. Geleneksel jilet ve elektrikli tıraş makineleri ise ciltte tahrişe neden olabilir; özellikle yenidoğan ve bebeklerin ince cildinde jiletle kesim tercih edilmemelidir.
Kılların koparılması (cımbızla veya ağda ile) ağrılı işlemler olduğundan bebek ve küçük çocuklarda uygun değildir. Ağda sonrası kıl büyüme süresi iki ila altı hafta arasında değişir, ancak hipertrikozlu hastalarda kıllar çok daha hızlı uzayarak hafta içinde rahatsızlık vermeye başlayabilir. Ağartma (bleaching) ise, hidrojen peroksit ile kılların rengini açarak haftaya kadar daha az belirgin hale getirir; açık tenli çocuklarda daha etkilidir, ancak tahrişe neden olabilir. Kimyasal tüy dökücüler cilde zarar verebilecek yüksek tahriş potansiyeline sahiptir ve yaygın hipertrikozisi olan çocuklarda sistemik toksisite riski nedeniyle kesinlikle kullanılmamalıdır.
Kalıcı ve Yarı Kalıcı Tedavi Yöntemleri
Elektroliz epilasyon oldukça etkilidir, ancak uzun seanslar, ağrı ve ciltte hiperpigmentasyon riski nedeniyle çocuklarda tolere edilebilir bir uygulama değildir.
Lazer ve yoğun darbeli fototedavi, mevcut en kalıcı epilasyon sistemleri olarak görülmekle birlikte, çocuklarda güvenilirliği ve etkinliği konusunda kapsamlı çalışmalar bulunmamaktadır ve genellikle yaş üstünde güvenli kabul edilir.
Topikal eflornitin krem kullanımı, ornitin dekarboksilaz enzimini engelleyerek kıl büyümesini yavaşlatır. Lazer tedavisi ile birlikte kullanıldığında etkinliği artar, ancak yaş altı çocuklarda ve yaygın hipertrikoziste güvenliği henüz tam olarak belirlenmemiştir.
Özet Tedavi Önerileri
-
Yaygın hipertrikozis için yaş altındaki çocuklarda modern elektrikli tıraş makineleri ve bariyer kremi, yaş ve üzeri çocuklarda ise tekrarlayan tıraşlar tercih edilebilir.
-
Lokalize hipertrikozis için yaş altındaki çocuklarda tekrarlayan tıraşlar, yaş ve üzeri çocuklarda ise tekrarlayan tıraş, kimyasal tüy dökücüler veya eflornitin hidroklorür kremi düşünülebilir.


