
- Gösterim: 54575
Sedef hastalığının nedenleri halen tam olarak açıklanamamakla birlikte genetik geçilişli olduğunu ve inflamasyonun yanlış düzenlemesinden kaynaklanan bir hastalık olduğu yönünde kanıtlarımız bulunmakta.
Sedef hastalığının genetik geçişinde 8. kromozom üzerinde PSORS I-VIII olarak tanımlanan gen lokasyonlarının sorumlu olduğunu biliyoruz. Bunların içerisinde HLA-Cw6 allel geni - PSORS I ana major gen lokasyonu. Evet genetik geçişli ancak hastalığını klinik tablosundan bir çok dışsal faktörün sorumlu olduğunu; ilaçlar, fiziksel sürtünme ve travmalar, enfeksiyon ve stres, görmekteyiz.
Geçmişten beri sedef tedavisinde kullandığımız methotrexate, cyclosporine (CyA), biyolojik ajanlar, immunotoxinlerin başarılı sonuçları sedef hastalığının immünolojik nedenlerini desteklemektedir. Ayrıca bu immünolojik neden desteğinde bazı sitokonlerin interferon alfa, beta, gama, interleukin (IL)-2; granülosit koloni stimüle eden faktör, bakteriyel süper antijenlerin sedef klinik tablosunu arttırması da yer almaktadır. Sedef klinik tablosunun iyileşmesi sırasında lezyonlarda T lenfositlerin, dermal dendritik hücrelerin, Langerhans hücreleri, nötrofiller ve bunlar tarafından yapılan TNF-a-, interferonlar ve IL-12/23 azalmaktadır. Bu tüm knıtlar sedef hastalığının deri ve ekleme spesifik immün sistem kaynaklı olduğunu göstermektedir.
Sedef hastalığında tüm tedavi protokollerinin temeli bu immün sistem kaynaklı deri ve hatta sedeften kaynaklanan eklemdeki inflamasyonun azaltılması üzerine kuruludur.
Sedef hastalığında biyolojik ajanlar tanımı aslında T lenfositleri ve inflamasyondan sorumlu sitokinleri baskılayan biyolojik ilaç protokollerini kapsamaktadır.
Tümör nekroz faktörü-alfa (TNF-α) psoriasis gelişiminde temel rol oynayan bir sitokindir. Günümüzde kullanılan biyolojik ajanların büyük bir bölümü bu sitokinin baskılanması üzerinedir ve anti TNF ajanlar olarak tanımlanmaktadır.
Biyolojik ajanlar; sistemik ilaçlar, fotokemoterapi gibi sistemik tedavilere cevap vermeyen, bu tedavilerin kullanılamadığı veya hasta tarafından kullanımı tolere edilemediği orta ve şiddetli plak psoriasis, stabil olmayan psoriasis ve psoriatik artritli yetişkin hastaların tedavisinde kullanılırlar.
Bir çok biyolojik ajan günümüzde seçenekler arasında sunulmakla birlikte ülkemizde anti-TNF ajanlardan; adalimumab ve infliksimab, reseptör blokeri olarak etanersept, anti(interlökin-12/23) monoklonal antikoru olarak ustekinumab aktif maddeleri psoriasis tedavisinde onaylıdır.
Biyolojik ajanların psoriaiste kullanımı için uygunluk ölçütleri, biyolojik ajanlar ile tedaviye uygun psoriasis hastalarının tanımları dünyada yapılmıştır. Ülkemizde de genel kabul görmüş olan bir değerlendirme ölçütüdür.
Biyolojik ajan tedavisi için uygunluk ölçütünde psoriasis hastalarının tanımlanması;
a. Orat-şiddetli psoriasis varlığında; PASI skoru ≥10, Dermatoloji yaşam kalitesi indeksi-DYK‹ ≥10 ve Vücut yüzey alanı VYA ≥%10 ve aşağıdaki durumlardan b ve ce nin valığı.
b. Klinikte hızlı kötüleşme, görünür alanların tutulması, fonksiyonel yetersizlik (palmoplantar, genital tutulum gibi), ağır eritrodermik veya jeneralize püstüler psoriasis, iş görmezliğini etkileyecek tırnak tutulumu, psöriatrik artit varlığı
c. En az 12 hafta süre ile asitretin metotreksat, siklosporin, fototerapi gibi sistemik tedavi kullanımına yanıtsız hastalarda, bu tedavilerin kontrendike olduğu yada kullanımı sırasında yan etki gelişen hastalarda, bu tedaviler devam ederken yada tedavi bitiminden sonra 3 ay içerisinde klinik tekrarlarda biyolojik ajanlar düşünülmelidir.
d. Yaşamı tehdit edecek eritrodermik veya jeneralize püstüler psoriasis varlığı. Bu madde yanlız başına biyolojik kullanımı için yeterli değildir.
Biyolojik Ajanların Tedavi Öncesi
Bu tedavilere başlanmadan önce hastalarda olası risk faktörlerini saptamak için ayrıntılı bir hastalık öyküsü-anamnez, fizik muayene, daha önce kullandığı ilaçların değerlendirilmesi ve gerekli laboratuvar tetkiklerinin göz önünde bulundurulması gerekmektedir.
- Psoriasisin tipi, süresi ve seyri, artrit varlığı sorgulanmalıdır.
- Psoriaiste daha önce kullandığı tedaviler, dozları, süreleri, varsa yan etkileri, etkinlikleri ve varsa ilacın kesilme nedenleri sorgulanmalıdır.
- Varsa eşlik eden diğer hastalıklar, düzenli olarak kullandığı ilaçlar sorgulanmalıdır.
- Psoriasisin hastanın yaşam kalitesine etkisi sorgulanmalıdır.
- Hastanın boy-kilo ölçümleri yapılmalı, vücut kitle indeksi hesaplanmalıdır.
- Anamnezde akut ve kronik enfeksiyonlar, tüberküloz (TB), kendisinde ve ailesinde, 1. derecede akrabalarında demiyelinizan hastalık ya da kanser olup olmadığı sorgulanmalıdır.
- Kadın hastalar gebelik açısından sorgulanmalı, şüpheli olgularda gebelik testleriyle gebelik dışlandıktan sonra biyolojik tedavi başlanmalıdır.
Biyolojik ajanların kullanımı sırasında
- Hastalar, ilk üç ay ayda bir, sonra üç ayda bir detaylı fiziki muayene ile kontrol edilmelidir.
- Tedaviye devam kararı, ortalama 12. haftada değerlendirilerek verilmeli, yanıt varsa devam edilmeli, ardından üç ayda bir izlenmelidir.
- Laboratuvar testleri; tam kan sayımı, tam idrar testi, karaciğer fonksiyon testleri, hepatit tarama testleri, C-reaktif protein, anti-HIV testi, gebelik testi, akciğer filmi, öncelikle interferon gama salınım testi (İGST; quantiferon tbc testi), yapılamıyorsa tüberkülin deri testi, protein purifiye derivate (TDT, PPD) yapılmalıdır.Tedavi sırasında da bu tetkikler belli aralıklarla ve gerekli hallerde daha sık aralıklarla tekrarlanabilir.
Biyolojik ajanların kullanılamayacağı durumlar -kontrendikasyonları
- Aktif enfeksiyonlar; bu ilaçların kullanımı öncesi ve tedavi sırasında hastalar periyodik olarak sistemik veya lokal enfeksiyonlar açısından değerlendirilmelidir. Aktif enfeksiyon tespit edildiği tedaviye arar verilmelidir. Bu ilaçların kullanımı sırasında salmonella ve listeriya enfeksiyonlarına karşı da dikkatli olunmalıdır.
- Aktif Tüberküloz-TB; ülkemizde tüberküloz malesef halen oldukça sık görülmekte olan bir enfeksiyondur. Bu nedenle biyolojik ajan kullanacak hastalarda TB açısından değerlendirme anamnez, fizik muayene, akciğer filmi, PPD testi (TDT testi) ve spesifik interferon-gama (quantiferon tbc testi) (İGST) analizi yapılmalıdır. Akciğer filminde TB sekelinin olması, akciğer TB olan bir hastayla son bir yıl içinde yakın temas içinde bulunulması (aynı oda havasını 24 saat boyunca solumak), TB açısından yüksek riskli sağlık personeli olunması, tedavi öncesi yapılan spesifik interferon-gama (quantiferon tbc testi) testinin pozitif veya ilk TDT değerinin ≥5 mm olması durumlarında ilk olarak tübörküloza karşı koruyucu tedavi yapılmalıdır. Koruyucu tedavi biyolojik tedaviden bir ay önce başlamalı ve herhangi bir nedenle biyolojik tedavi kesilse bile 9 ay boyunca 300 mg/gün izoniazid (İNH) verilmelidir. İNH’nin kullanamadığı durumlarda 4 ay boyunca rifampisin kullanılabilir. Biyolojik tedavi alan hastaların her 3 ayda bir ve tedavi kesildikten sonra 6 ay sonrasına kadar TB açısından klinik olarak izlenmesi önerilmektedir. Ayrıca TB’nin sadece akciğer değil, akciğer dışı organ tutulumu, atipik ve dissemine formları da dikkate alınmalıdır. Türkiye’de anti-TNF tedavi ve ustekinumab uygulanan hastalar için üç ayda bir “Güvenlik İzlem Formu” doldurulması ve Sağlık Bakanlığı’na iletilmesi zorunludur. TB için yılda bir akciğer grafisi ve gerektiğinde quantiferon tbc testi ile hasta izlenir. Eğer bu tetkiklerde şüpheli durumlar oluşursa tomografi vs. gibi daha detaylı tetkikler yapılmalıdır. Ayrıca rutin kontrollerde sebebi bilinmeyen ateş, gece terlemeleri konusunda hekim uyanık olmalıdır.
- İmmünosüpressif tedavi
- Kanser varlığı; Biyolojik tedaviye başlamadan önce hastalar mutlaka malignite açısından değerlendirilmeli, bu amaçla dikkatli öykü alınmalı ve detaylı fizik muayene yapılmalıdır. Öncesinde psoriais yada başka nedenle 200 seans PUVA, 350 seans UVB ile iki yıldan fazla aralıksız siklosporin almış olan hastalarda malignite riski artmış olduğundan daha dikkatli olunmalıdır. Ailesel malignite öyküsü olan hastalarda da yine daha dikkatli olunmalıdır. Tedavi öncesinde 5 yıldan uzun kür sağlanmış solid organ tümörü, melanom dışı deri kanseri olan olgularda biyolojik ajan kullanılabilir. Biyolojik tedavi kullanan psoriasisli hastalar her vizitte öykü ve detaylı fizik muayene ile özellikle lenfoma ve diğer maligniteler açısından izlenmelidir. Biyolojik ajan ile tedavi sırasında malignite gelişirse tedavi derhal kesilmelidir.
- Demiyelinizan hastalıklar; Biyolojik tedavi ile demiyelinizan hastalıkların gelişimi arasında bir ilişki olabileceğinden biyolojik ajanlar, multipl skleroz (MS) ve diğer demiyelinizasyon hastalıklarında kullanılmamalıdır. MS’li hastaların birinci derece akrabaları da MS gelişimi için artmış riske sahip oldukları için bunlarda da bu ajanlar kullanılmamalıdır.
- Konjestif kalp yetmezliği [New York Kalp Birliği (NYKB) derece 3 ve 4]; Orta ve şiddetli konjestif kalp yetmezliğinde (NYKB 3-4 derece) antiTNF ajanlar kontrendikedir. Hafif konjestif kalp yetmezliğinde (NYKB 1-2 derece) dikkatli kullanılmalıdır. Ekokardiyografi ile izlenmeli, ejeksiyon fraksiyonu %50’nin altında ise verilmemelidir. Yeni semptom gelişimi veya semptomların kötüleşmesi durumunda tedavi kesilmelidir.
- Biyolojik ajana karşı hastanın duyarlılığı-hipersensitivite.
- HIV pozitif veya AIDS’li hastalar
- Hepatit B veya C pozitif hastalar; Biyolojik ajan tedavisi başlamadan önce hepatit B ve C taraması yapılmalıdır. Eşzamanlı hepatit B ve C enfeksiyonu olan hastalarda anti-TNF tedavi verilmemesi gerektiği yönünde bir Amerikan Gıda ve İlaç Dairesi uyarısı mevcuttur. Hepatit C virüs enfeksiyonu varlığında düzenli takiple tedavi verilebilirken, Hepatit B virüs enfeksiyonu varlığında kullanım zorunlu ise, karaciğer fonksiyon testleri yanında ek olarak viral yükün de tedaviden önce, tedavi sırasında ve tedaviden üç ay sonrasına kadar izlenmesi gerektiği bildirilmiştir.
- Gebelik ve emzirme;Anti-TNF ajanlar ve ustekinumab için gebelik kategorisi B’dir. Ancak gebelik ve laktasyon döneminde biyolojik ajan kullanımı ile ilgili klinik çalışmalar mevcut olmadığı için bu hastalarda biyolojik ajan kullanımı önerilmemektedir. Gebe kalma potansiyeli olan kadınlarda biyolojik tedavi sırasında ve sonrasında 6 ay boyunca korunma uygulanmalı gebeliğe izin verilmemeldir. Biyolojik tedavi altında iken gebelik saptanırsa ilgili uzmana yönlendirilerek daha detaylı değerlendirme yapılmalı; biyolojik tedavinin devamı konusunda birlikte karar verilmelidir. Bu durum, yarar/zarar oranı dikkate alınarak hasta bazında değerlendirilmelidir. Gebelikte anti-TNF kullanan annelerin bebeklerinde aşıya bağlı ciddi enfeksiyonlar görülebileceğinden, bebeklerde Basil Calmet Guerrin dahil canlı aşıların yapılması 6. aydan sonraya ertelenmelidir. Biyolojik ajanların anne sütüne geçişi konusunda kesin bilgiler olmamakla birlikte yarar/zarar oranı gözetilerek emzirme durumunda ilacın kullanımı sürdürülebilir. Biyolojik ajanların sperm kalitesine etkisi bulunmadığından erkek hastalarda korunma gerekmez.
- Aşılanma; aşılanmaya ihtiyaç duyulan psoriasis hastalarında tüm aşılar biyolojik tedaviye başlamadan en az 15 gün önce yapılmalıdır. Biyolojik tedavi adayı hastalarda tedaviden önce pnömokok ve influenza aşılarının yapılması önerilmektedir. Biyolojik tedaviye başlandıktan sonra canlı aşılar uygulanmamalıdır.
- Kronik Böbrek Yetmezliği; yeterli veri olmamakla birlikte güncel literatür bilgileri ışığında zorunlu durumlarda böbrek yetmezliği olan hastalarda biyolojik ajanların dikkatli bir izlemle kullanılabileceği değerlendirilmektedir.
- Cerrahi Müdahale; büyük bir ameliyat durumunda biyolojik ajanlar geçici olarak kesilir, hasta normale döndükten sonra tedaviye tekrar başlanabilir. Yine acil cerrahi müdahalelerde ajan kesilerek cerrahi operasyon yapılır, hasta normale dönünce tedaviye tekrar devam edilir. Majör yanık ve travmalarda biyolojik ajanlar hasta normale dönene kadar kesilmelidir. Minör yanık ve travmalarda ise, tedaviyi kesmeye gerek yoktur. Diş çektirmek veya diş tedavisi için; müdahale majör bir girişim ise, ilaç kesilmeli, minör bir girişim ise ilacın kesilmesine gerek olmadığı değerlendirilmektedir. Bu durum klinisyenin görüşüne bağlı olarak hasta bazında değerlendirilmelidir.
- Çocuklarda ve Yaşlılarda Kullanımı; günümüzde ülkemizde çocuk yaşta psoriasiste onaylı tek biyolojik ajan etanersepttir. Adalimumab ise onay aşamasındadır. Diğer ajanların bu yaş grubundaki kullanımları henüz onamlı değildir. Biyolojik tedaviler için psoriasiste bir üst yaş sınırı olmamakla birlikte bu ilaçların yaşlı hastalarda kullanımı ile ilgili çalışmalar henüz mevcut değildir.
- İlaç Etkileşimi; bu ajanlarla birlikte glukokortikoidler, nonsteroid antienflamatuvar ilaçlar, analjezikler ve metotreksat kullanıldığında etkileşim
bildirilmemiştir. Ancak ciddi enfeksiyon ve nötropeni riskinden dolayı anti-TNF ajanlarla birlikte ialça kulanımında dikkatli olunmalıdır.
Biyolojik ilaçların sedefte kullanımı herbiri için ayrıca geliştirilmiş bir takip protokollü içermektedir. Bu protokoller değişmekle birlikte hastalardan genel olarak; karaciğer fonksiyon testleri, tam kan ve trombosit sayımı, hepatit ve tüberküloz panel taraması istenmekte ve tedavi boyunca bunlar tekrarlanmaktadır.
Sedef hastalığında kullanılan biyolojik ilaçları aşağıdaki gibi guruplarda toplayabiliriz.
- Asıl hedefleri patojenik T lenfositler üzerinden olanlar
- TNF alfa üzerinden olanlar
- IL-12 ve Il-23 üzerinden olanlar
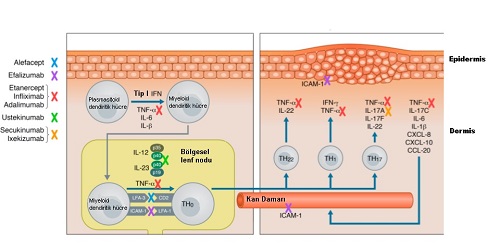
Patojenik T lenfositler üzerinden etki gösteren biyolojik ilaçlar
Alefacept
Alefacept rekombinasyon tekniği ile geliştirilmiş bir protein. IgG antikorları Fc kısımları ile T lenfositleri yüzeyindeki CD2 proteinlerine bağlanarak T lenfositlerde inflamatuvar reaksiyonu başlatmakta. Alefacept memory T lenfositler üzerindeki bu CD2 proteine bağlanarak bu hücreleri IgG arafından aktivasyonunu baskılamaktadır. 2003 yılında kullanımına izin verilmiş ancak 2011 yılında ilaç kulanımdan çekilmiştir.
Efalizumab
LFA-1'in CD11 alt ünitesine karşı hazırlanmış insan monoklonal antikorudur. 2003 yılında kullanımına izin verilen ilaç 2009 yılında kullanımı sırasında multifokal lökoansefalopati olgularının ortaya çıkmasından sonra kullanımdan çekilmiştir.
TNF alfa üzerinden etki göstern biyolojik ilaçlar
İnfliksimab
Tümör nekroz faktörü-alfa (TNF-α) antagonistidir. Kimerizim (fare/insan) elde edilen bir monoklonal antikordur. TNF-α ye spesifik ve yüksek oranda afinite gösteren bu antikor TNF-α inhibe ederek bunun rol oynadığı inflamasyonu baskılamaktadır. Bu nedenle psoriaisis ve inflamtuar cilt hastalıklarında tedavide kullanılmaktadır.
TNF-α'yı bloke etmesi ve stabil kompleksler oluşturmasının yanı sıra, hücre membranındaki TNF-α’ya bağlanarak antikor aracılı ile TNF-alfa üreten hücrelerin öllümünüde sağlamaktadır.
Bu etkilerini hızlı ve seçici yapmakta ve seçici etkileri nedeni ile selektif immünsüspresif ajanlar grubunun bir üyesi olarak sınıflamdırılır.
Sıradan psoriasis tedavilerine yanıt vermeyen, bu tedavilerin kullanılamadığı veya hasta tarafından tolere edilemediği orta-şiddetli psoriasis ve psoriatik artritli yetişkin hastaların tedavisinde kullanılır.
İnfliksimab ülkemizde Remicade 100 mg Konsantre IV İnfüzyon liyofilize flakon ve Remsima 100 mg IV EMSIMA 100 MG I.V. İnfüzyon liyofilize flakon şeklinde onamlı olarak kulanılmaktadır.
Flakon şişelerinde 2-8 C arasında saklanmalıdır.
Hastaya uygulanacak doz 250 ml serum fizyolojik içerisinde hazırlandıktan sonra bir filtre sisteminden geçirilerek IV olarak verilir. Solüsyon hazırlandıktan sonra hemen veya en fazla üç saat içerisinde uygulamaya başlanılması gerekmektedir. 2 saatin üzerinde yavaş infüzyonla verilir.
Genel doz şemesı hasta kilosuna göre kilogram başına 5 mg olarak hesaplanır.
0, 2 ve 6. haftalarda 3 doz uygulandıktan sonra her sekiz haftada bir tekrarlanan idame dozlar uygulanır. Tedavi sırasında sedefin klinik yanıtına göre doz aralıkları düzenlenebilir.
İnfüzyon sonrası ilacın vücutta eliminasyon yarılanma zamanı 8.5-9 gündür. Ancak doz ve kullanım süresine bağlı olarak 28 haftaya kadar vcutta tespit edilebilmektedir.
İlacı uygulanması esnasında veya infüzyonu takip eden bir saat içerisinde görülen yan etkileri kapsayan reaksiyonlar tedavinin kesilmesi için en sık görülen sebeptir. Bu reaksiyonlar hastaların yaklaşık %18’inde ortaya çıkmaktadır. İnfüzyon reaksiyonları genellikle 2. veya 3. uygulamada infüzyonda ortaya çıkar. İnfüzyon reaksiyonu ilk 24 saat içerisinde ortaya çıkan akut reaksiyon şeklinde görülebildiği gibi, infüzyondan sonraki 24 saat ile 14 gün arasında görülebilen gecikmiş tip aşırı duyarlık reaksiyonu şeklinde de olabilir. Düzenli aralıklarla idame tedavisi alan hastalarda infüzyon reaksiyonu gelişme riski daha azdır. Plak tip psoriasisli hastalarda infüzyon reaksiyonu gelişme riskinin daha az olduğu (%10) düşünülmektedir. İnfliksimaba karşı nötralizan antikor gelişme riski yaklaşık %10-30 arasında olup, antikor gelişen hastalarda reaksiyon riskinin daha fazla olduğu bilinmektedir.
İnfliksimab seçici immünosüpresif bir ilaç olarak tanılanmakla birlikte tedavisi esnasında enfeksiyon gelişme veya mevcut sessiz bir enfeksiyon aktivasyon riski, tüberküloz ve diğer fırsatçı enfeksiyonlarda aktivasyon riski, multipl skleroz ve diğer demiyelinizasyon hastalıklarının gelişmesi veya kötüleşmesi, alerjik reaksiyonlar, otoantikor gelişimi, konjestif kalp yetmezliğinin ağırlaşması, alanin aminotransferaz ve aspartat aminotransferaz düzeylerinde artış, lenfoma ve başta deri kanserleri olmak üzere diğer maligniteler nadir olarak görülebilir.
İnfliksimab tedavisi alanların %50 kadarında antinükleer antikorlar gelişebilir ve genellikle geçici yapıdadır. Bu antikorlar genellikle klinik bulguya sebep olmaz ancak çok nadiren genellikle deriye sınırlı lupus-benzeri sendrom gelişebilir ve tedavinin kesilmesi gerekebilir.
İnfliksimabın anne sütüne geçişi konusunda deliller sınırlı olmakla birlikte emzirme önerilmez.
İnfliksimabın çocuk hastalarda kullanımı ile ilgili deneyimler olgu takdimleri ile sınırlıdır. Bu konuda yapılmış kontrollü çalışma mevcut değildir. Hekim, pediatrik kullanım için fayda/risk oranını gözeterek olgu bazında kendi kararını vermelidir.
Siklosporin ile beraber kullanımında immünsüpresyonda artışa olabilme riskinde dikkatli olunmalıdır.
İnfliksimabın düşük doz metotreksat ile kombinasyonu uzun dönem etkinliğini korumak için önerilir. Ancak bu durumda daha dikatli bir takip gerekmektedir.
Retinoidlerle beraber kullanımına dair deneyimler sınırlıdır.
Psoralen-ultraviyole A ile kombinasyonu PUVA deri kanseri riskini artırabilir.
Başka bir biyolojik ile birlikte kullanımı enfeksiyon riskini artırdığı için önerilmez.
Topikal steroidler ve toikal vitamin D3 analogları gibi ilaçlarla gerektiğinde kombine edilebilir.
Psorisis normal klinik tablosu dışında psorisis tırnak tutulumundada etili olduğu gösterilmiştir.
Psoriaista eklem tutulumunda, etkinliği artırmak amacı ile ve antikor gelişim riskini azaltmak için tedaviye düşük doz metotreksat eklenebilir.
Adalimumab
TNF-alfa antagonistidir. Tamamı insandan elde edilen monoklonal antikordur. Adalimumab spesifik olarak TNF-alfa ile p55, p75 hücre-yüzey reseptör ilişkisini bloke ederek, TNF-alfa’nın biyolojik aktivitesini baskılar. Bu nedenle psoriaisis ve inflamtuar cilt hastalıklarında tedavide kullanılmaktadır.
Adalimumab, fototerapi ve geleneksel sistemik tedavilere yanıt vermeyen veya bu tedavilerin kullanılamadığı-kontrendike yada hasta tarafından tolre edilemediği orta ve şiddetli kronik plak tip psoriasis tedavisinde kullanılmaktadır. Avrupa İlaç Ajansı topikal tedavi ve fototerapiye yanıt vermeyen veya bu tedavilerin kontrendike olduğu 4 yaş ve üzeri şiddetli kronik plak tip pediatrik psoriasis tedavisi için onay vermiştir.
Tedavi sırasında kanda regülatör T (Treg) lenfosit sayısının arttığı, buna karşılık hafıza (memory) B lenfosit sayısının değişmediği gösterilmiştir. Bunun nedeni adalimumabın lenfotoksin (TNF-beta) blokajı yapmamasına bağlanmıştır.
Ülkemizde adalimumab Amgevita 40 mg/0.8 ml kullanıma hazır enjektör(2 adet), Humira 20mg/0.2 ml ve 40 mg/0.4 ml kullanıma hazır enjektör onamlı olarak kullanılmaktadır.
Adalimumab 0,5 mg/kg dozlarda kullanılmaktadır. Bu tek doz sonrası vücutta yarılnama süresi 11,8 gün olduğu gösterilmiştir.
Adalimumab deri altı- subkutan enjeksiyonlar şeklinde uygulanmaktadır.
Psoriasis tedavisinde ilk uygulamada 80 mg ve 1 hafta sonra 40 mg ile yükleme yapılır ve daha sonra tedavi düzenli aralıklarla 2 haftada bir 40 mg subkutan adalimumab uygulaması ile sürdürülür. Aşırı kilolu, obez hastalarda bu sabit doz uygulaması geçerlidir.
Etkinliği 12. haftada değerlendirilir.
Palmoplantar psorisiste de etkilidir.
Tedavi sırasında en sık gözlenen yan etki enjeksiyon bölgesindeki ağrı ve eritemdir. Sık gözlenen diğer yan etkiler ise üst solunum yolu enfeksiyonları, sinüzit, baş ağrısı ve morbiliform deri döküntüleridir. Ürtiker, transaminazlarda yükselme, püstüler dermatit gelişimi, kaşıntı, anjiyoödem, trombositopeni, lökopeni nadiren görülür. Konjestif kalp yetmezliğinde kötüleşme, malignite riskinde artış ve tüberküloz reaktivasyonu çok nadir olarak bildirilmiştir.Tedavi sırasında ortaya çıkan yan etkiler daha çok yaşlı hastalarda görülmekte ve daha ciddi seyretmektedir.
Gebelerde ve emzirme döneminde kullanımı önerilmemektedir.
Psoriasis ve hepatit C birlikteliğinde adalimumab tedavisi için kontrendike bir durum söz konusu değildir ancak bir gastroenteroloji uzmanı eşliğinde takip ve tedavi süresince viral yük monitorizasyonu gereklidir.
Adalimumab diğer anti-TNF’ler, anakinra (interlökin-1R antagonisti) ve abatacept ile birlikte ciddi enfeksiyon riskini artırdığından kullanılmamalıdır.
Adalimumab Avrupa’da, Avrupa İlaç Ajansı tarafından kronik plak tip pediatrik psoriasis tedavisi için onaylanmıştır. Çocuklarda 1 hafta ara ile 0,8 mg/kg (maksimum 40 mg olacak şekilde) 2 kez ve daha sonra 2 haftada bir aynı dozda subkutan olarak devamı önerilmektedir. Kombinasyon Tedavileri Adalimumab ile kombinasyon tedavisi için kontrollü çalışmalar olmamakla birlikte düşük doz (7,5-10 mg/hafta) metotreksat ile birlikte kullanıldığında etkinliğin arttığı ileri sürülmektedir.
İnterlökinler üzerinden etki gösteren biyolojik ajanlar
Ustekinumab
Etki mekanizması Il-12 ve Il-23 üzerindendir. Tamamen insandan elde edilen ve interlökin-IL-12 ve Il-23 sitokinlerin ortak p40 alt birimlerini yüksek spesifisite ve benzerlikle bağlayan Ig G antikorudur. Bu etkileri ile psoriaiste ve inflamatura hastalıklarda kullanılmaktadır.
Ustekinumab, Amerikan Gıda ve İlaç Dairesi ve Avrupa İlaç Ajansı tarafından psoriasis için 2009’da, psoriatik artrit için 2013’te ve 12 yaş ve üstü erişkin psoriasis tedavisi için 2015’te onay almıştır. Ülkemizde ise 2013 yılından itibaren orta-şiddetli psoriasis ve psoriatik artrit tedavisinde onaylıdır ve STELARA 45 mg, 90 mg ve 130 mg SC kullanıma hazır enjektör formları bulunmaktadır.
Ustekinumab etki mekanizmasında İL-12 ve İL-23 baskılaması bunların Th1 ve Th17 hücrelerinden tümör nekroz faktörü (TNF)-alfa, interferon-gama, İL-2, 6, 17, 21, 22 salınımı da baskılanmış olur.
Kronik plak psoriasiste 45 mg, 100 kg üzeri hastalar için ise 90 mg dozunda kullanılır. 0. ve 4. haftalarda bir enjeksiyon, sonrasında 12 haftada bir enjeksiyon deri altı-subkutan olarak yapılır.
Ustekinumabın avantajı 12 haftada bir (ilk yıl yılda 5 kez, daha sondaki yıllarda yılda 4'er kez) subkutan yolla uygulanabilen bir tedavi seçeneği olmasıdır.
Tedavi öncesi ve tedavi sırasında; tam kan sayımı, karaciğer fonksiyon testleri, viral serolojik tetkikler, anti-HIV, gebelik testi yapılmalı ve akciğer grafisi çekilmelidir.
Ustekinumabın yan etkileri, anti-TNF ajanlarla benzerdir. Sık rastlanan yan etkiler; üst solunum yolu enfeksiyonları, nazofarenjit, artralji, öksürük, baş ağrısıdır. Nadiren enjeksiyon yeri reaksiyonları (%1-5) ve nötralizan antikor gelişimi (%5) gözlenir. Malignite ve nonmelanoma deri kanserleri gelişimi olgu bildirileri ile sınırlıdır. Ustekinumab ile tedavi edilen hastalarda bildirilen bu malignite insidansı, genel popülasyonda beklenene yakındır. Dört yıllık süre ile ustekinumab kullanımı ile majör kardiyovasküler olaylarda (MACE) genel psoriatik popülasyona kıyasla bir benzerlik görülmektedir.
Enfeksiyon ve latent enfeksiyon reaktivasyon riskini artırır. Hastalar, şiddetli ve atipik enfeksiyonlar açısından yakından izlenmeli ve şüpheli durumlarda tedavi kesilmelidir.
Ustekinumabın kombinasyon tedavisi için kontrollü çalışma yoktur. Topikal ajanlarla kombine edilebilir.
Ustekinumabın pediatrik kullanımı ile ilgili herhangi bir veri mevcut değildir.
Sekukinumab
İnterlökin (İL)-17A’ya selektif olarak bağlanarak nötralize eden, immünoglobulin G1/κ izotipinde rekombinan, yüksek afiniteli, insan monoklonal antikorudur.
İL-17A’yı hedef alarak ve keratinositler dahil çeşitli hücre tiplerinde salgılanan İL-17 reseptörü ile etkileşimini inhibe ederek etki gösterir.
Gleneksel sistemik tedavilere yanıt vermeyen veya bu tedavilerin kontrendike olduğu orta ve şiddetli kronik plak tip psoriasis tedavisi için endikedir. Bu endikasyon için 2015 yılında Amerikan Gıda ve İlaç Dairesi ve Avrupa İlaç Ajansı tarafından onay almıştır. Ülkemizde VERXANT 150 mg SC enjeksiyon için liyofilize toz içeren flakon olark kullanılmaktadır.
Önerilen doz deri altı-subkutan enjeksiyon yoluyla 300 mg sekukinumab olup 0, 1, 2 ve 3. haftalarda yapılmakta bunu takiben 4 haftada bir aylık idame dozu şeklinde uygulanır. Her 300 mg doz, 150 mg’lik iki subkutan enjeksiyon halinde verilir.
16 haftaya kadarki sürede yanıt vermeyen hastalarda tedavinin kesilmesi düşünülmelidir.
Sekukinumab tedavisi 12-16. haftalarda değerlendirilmektedir. Etkinlik devam ettiği ve yan etki gelişmediği sürece tedaviye devam edilebilir.
En sık bildirilen yan etkiler; üst solunum yolu enfeksiyonu (ÜSYE,nazofarenjit, rinit) olmuştur. Yaygın olarak görülen diğer yan etkiler herpes labialis, rinore ve diyaredir. Yaygın olmayan yan etkiler ise oral kandidiyazis, tinea pedis, otitis eksterna, nötropeni, konjonktivit ve ürtikerdir.
Ciddi enfeksiyonlar, sekukinumab uygulanan hastaların %0,14’ünde görülmüştür. Nötropeni, plaseboya kıyasla sekukinumab ile (%0,5) daha sık gözlenmiştir, fakat olguların çoğu hafif, geçici ve geri dönüşümlü olmuştur.
Aktif Crohn hastalığı olan hastalara sekukinumab reçete edilirken dikkatli olunmalıdır. Klinik çalışmalarda gerek sekukinumab gerekse plasebo gruplarında, Crohn hastalığında şiddetlenmeler gözlenmiştir.
Klinik çalışmalarda sekukinumab kullanan hastalarda nadir anafilaktik reaksiyon olgusu gözlenmiştir. Anafilaktik ya da başka bir ciddi alerjik reaksiyon ortaya çıkarsa, sekukinumab uygulaması derhal durdurulmalı ve uygun tedavi başlatılmalıdır.
İmmünojenisite, sekukinumab ile tedavi edilen hastaların %1’inden azında görülmüştür. Anti-ilaç antikorlarının yaklaşık yarısı nötralizan yapıdadır, ancak bu durum etkililik kaybı ya da farmakokinetik anormallikler ile ilişkilendirilmemiştir1. Gebelik kategorisi B olarak belirtilmektedir.
Canlı aşılar, sekukinumab ile eşzamanlı uygulanmamalıdır. Sekukinumab alan hastalar eş zamanlı olarak inaktive ya da canlı olmayan aşıları alabilir.
Psoriatrik artrit ve ankilozan spondilitli hastalarda sekukinumabın metotreksat ve/veya kortikosteroidler ile eş zamanlı kullanımında herhangi bir etkileşim saptanmamıştır.
Ixekizumab ve Brodalumab
Etki mekanizması sekukinumab benzemekte.
Ülkemizde henüz yok.
Faz çalışmaları henüz bitmemiş olanlar ise; tildrakizumab, risankizumab, guselkumab.
Biyolojik ajan kullanımında tartışma konusu; Biyolojik ajalnların biyobenzerleri
Öncelikle biyobenzer ilaçlar, orijinal ilacın özdeşi değildir ve bu ilaçların kullanımı sonucu ortaya çıkacak etkinlik ve yan etkiler henüz tam olarak bilinmemektedir. Bunların farklı etki, yan etki ve güvenlik profilleri olması kaçınılmazdır. Ayrıca biyobenzerlerin adlandırma ve takiplerinin ne şekilde yapılacağı ve bu ilaçların birbiri yerine kullanılması ya da değiştirilmesinin nasıl olacağı henüz kesin olarak belirlenememiştir. Bu tür ilaçların birbirinden farklı adlandırılması gerekmektedir.
Dünya Sağlık Örgütü, biyobenzerlerin takip edilebilir olması için özgün kimlikleri olması gerekliliği üzerinde durmaktadır. Özellikle yan etki takibinde biyoteknolojik ilacın uluslararası patentsiz ismine “International Non-Proprietary Names” (INN) ek olarak marka adı, üretici firmanın ismi, lot numarası ve üretildiği ülke bilgilerinin de sağlanması istenmektedir.
Ayrıca referans ürünle biyobenzer ürünün prospektüslerinde ürüne özgü güvenlik, pozoloji, kontrendikasyonlar, uyarılar ve yan etkilerin de ayrı ayrı belitilmesi beklenmektedir.
Biyobenzer ilacın özgün bir isme sahip olması şu nedenlerle önemlidir: Herşeyden önce biyobenzer ürünler, adında da belirtildiği gibi referans ürünle özdeş değildir. Özdeş olmayan iki ilacın aynı ismi taşıması bu iki ilacın hatalı bir şekilde birbirinin aynı gibi kabul edilmesi ve otomatik olarak değiştirilebilmesi anlamına gelir ki bu durum ciddi sorunlar doğurabilir. Örneğin; birbirinin aynı olmayan iki ilacın etki ve yan etkilerinin takibi ancak bunların farklı isimlerle isimlendirilmeleri sayesinde gerçekleştirilebilir. Diğer türlü birinde ortaya çıkacak yan etki ya da etki azlığı haksız yere diğer ilaca da mal edilmiş olacak ve bu şekilde doğru bir takip gerçekleşemeyecektir.
Bunun dışında bu ilaçların farklı INN’ler ile adlandırılması ilacın reçetelenmesi, hastaya uygulanması ve doktorlar ya da bilim adamları tarafından ilaçla ilgili iletişim kurulup bilgi alışverişi yapılmasını da daha kolay bir hale getirecektir. Bir ilacın değiştirilebilir olması, tedavi devam ederken tedavide kullanılan bir ilaçtan hekimin insiyatifi ile eşdeğer olan başka bir ilaca geçilebilmesidir. Bu karar yalnızca hekim tarafından verilebilir. Yerine kullanılabilme ise hekim tarafından özellikle belirtilmediği sürece aynı INN ile adlandırılan ilaçların hekimin rızasına gerek duyulmadan eczane ya da hastanede birbirinin yerine verilebilmesidir. Değiştirilebilme ve yerine verilme durumları birçok kimyasal ilaç için mümkündür ve sıkça yapılmaktadır. Ancak biyoteknolojik ürünlerde durum farklıdır.
Avrupa İlaç Ajansı başta olmak üzere çeşitli Avrupa ülkeleri enstitü ve ajansları biyoteknolojik ilaçların hekimin onayı ile değiştirilebilir olabileceğine, hekimin insiyatifi dışında yerine kullanılamayacağına karar vermiştir. Çünkü referans ilaçla biyobenzer ilacın birbiri yerine kullanılabilmesi için bunların hem yapıca özdeş olması hem de biyolojik olarak kanıtlanmış eşitliklerinin olması gerekmektedir.
Aynı moleküler yapıya sahip olan ilaçların birbiri yerine kullanılabilmeleri kimyasal ilaçlar için mümkün olabilir. Çünkü bu durumda orjinal ve jenerik ilaçlar etkinliği aynı olan özdeş ilaçlar olarak kabul edilir ki bu ilaçlarda bile aynı kimyasal bileşik olmalarına rağmen bazı hastalar tarafından tolere edilebilememe durumu söz konusudur.
Birbirinden bağımsız olarak üretilen biyoteknolojik ilaçlar ise protein sekansı, moleküler yapısının katlanma şekli, glikolizasyon paterni, fonksiyon ve immünojenitesi açısından diğerinden farklıdır. Bu nedenle uygun düzenlenmiş geniş katılımlı klinik çalışmalarla desteklenmediği sürece bu ilaçlar birbiri yerine kullanılamaz. Aslında bir biyoteknolojik ürünün hekimin onayı dahilinde bile biyobenzeri ile değiştirilmesi kaçınılması gereken bir durum olmalıdır. Çünkü bir biyolojik üründen diğerine geçmek antikor üretim riskini ciddi şekilde arttırmaktadır. Bu nedenle uygun çalışmalardan geçmiş ve onaylanmış ürünler bile olsa bunların değiştirilerek birbiri yerine kullanılması uygun değildir.
Eğer bir değişiklik yapılacaksa bu durum hastanın tedavi sürecindeki bir değişiklik olarak kabul edilmeli ve bu değişim yalnızca kesin tıbbi nedenler varsa yapılabilmelidir. Eğer referans ilaç yerine biyobenzer ilaç kullanımı planlanıyorsa bunun tedavi başlamadan önce belirlenmesi ve o şekilde kullanılması daha uygun olacaktır. Bu şekilde hem etkinlik ve yan etkiler daha güvenli ve doğru olarak takip edilebilecek hem de biyobenzerle ilgili veri birikimi sağlanıp bu iki ürünün birbiri ile karşılaştırılması mümkün olabilecektir.
Biyobenzer ilaçların referans ilaçlar gibi güvenli olduğunu kanıtlayan yeterli veri bulunduğu durumlarda bile bu ürünlerin birçoğunun referans ilaca göre çok daha az hastada kullanılmış olduğu bilinmektedir. Ayrıca biyobenzer ilaçların onay alma süreci daha kısadır ve bu ilaçların etkinliği ve yan etkileri kısıtlı sayıda hastada denenmiştir. Bu nedenle özellikle belirli bir klinik tecrübenin henüz oluşmadığı biyobenzer ilaçlarla ilgili olarak bazı doktor ve hastalarda güvensizlik durumu söz konusu olabilmektedir.
Bu nedenle ilacın değiştirilebilir ya da yerine kullanılabilir olması, bu konuda endişesi olmayan hekimin insiyatifinde olmalıdır. Biyobenzer ilaç üretmek için o ürünün klinik araştırmaları dahil tüm araştırmalarının yapılması gereklidir. Biyobenzer ürünün klinik çalışmasının başka bir hastalık için yapılmış olması, biyobenzer ürünün o hastalığa ait klinik çalışması olduğu anlamını taşımaz. Biyobenzerlerin kullanımı ve takibi ile ilgili esasların sağlık otoritesi tarafından bir an önce belirlenmesi, bu ürünlerin birbirinden ayrımının sağlanması ve bu ilaçların birbirinin yerine kullanım ve değiştirilebilme yetkisinin hekime bırakılması hasta sağlığı ve takibi açısından uygun olacaktır.


